¶ Folha de rosto
© 2025 Ministério da Agricultura e Pecuária. Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial. A responsabilidade pelos direitos autorais de textos e imagens desta obra é do autor.
Elaboração, distribuição, informações:
Ministério da Agricultura e Pecuária
Secretaria de Defesa Agropecuária - SDA
Departamento de Serviços Técnicos - DTEC
Esplanada dos Ministérios, Bloco D, Anexo, Ala B, 4º andar, sala 433
CEP: 70043-900, Brasília - DF
www.agricultura.gov.br
e- mail: cgal@agro.gov.br
Central de Relacionamento: 0800 704 1995
Equipe técnica:
André Barbosa da Silva – SLAV-SC/LFDA-RS
Diego Cesar Pelolungo – LFDA-MG
Érico Silva Pires — LFDA-GO
Heitor Daguer — DIDEL/COSLAB/CGAL
Juarez Fabiano de Alkmim Filho – LFDA-MG
Lailah Nunes Santana Sampaio – COGETEC/CGAL
Michele Fabiane de Oliveira — LFDA-MG
Nélio Monteiro de Sousa – LFDA-PA
Ricardo Pimenta — SLAV-SC/LFDA-RS
Tiago Follmann Perin – LFDA-RS
Agradecimentos:
Carla Ivone Carraro – Comissão de Métodos Analíticos/Sindirações
Gilberto Batista de Souza - Empresa Brasileira de Pesquisa Agropecuária/Embrapa Pecuária Sudeste
¶ Folha resumo
|
Macroprocesso: Laboratórios |
Objetivo: Estabelecer os métodos oficiais de análise de alimentos para animais destinados ao uso nos laboratórios da Rede Nacional de Laboratórios Agropecuários. |
|||
|
Processo: Análises laboratoriais |
||||
|
Entrega: Segurança e qualidade de alimentos |
Público alvo e demais interessados: Laboratórios Federais de Defesa Agropecuária e laboratórios credenciados pelo Ministério da Agricultura e Pecuária (MAPA). |
Versão do documento: 7 |
||
|
Setor responsável e responsabilidades A Coordenação Geral de Laboratórios Agropecuários do Departamento de Serviços Técnicos é responsável pela elaboração, atualização e envio para aprovação deste manual, tendo responsabilidade quanto aos procedimentos descritos no documento. |
||||
¶ 1. Definições e conceitos
¶ 1.1. Definições
Níveis de garantia: são as informações obrigatórias sobre a qualidade nutricional de um produto destinado à alimentação animal, expressas em valores mínimos e/ou máximos, que guardam correlação com sua composição. Os níveis de garantia são expressos em mg/kg quando a concentração for inferior a 10.000 mg/kg ou em g/kg quando for igual ou superior a 10.000 mg/kg (10 g/kg). Outras unidades de medida dos níveis de garantia poderão ser empregadas, desde que aprovadas pelo MAPA.
¶ 1.2. Conceitos
Não aplicável
¶ 2. Responsabilidades
O presente manual possui vigência e prazo indeterminado e será revisado sempre que necessário e no mínimo anualmente pela Coordenação Geral de Laboratórios Agropecuários do Departamento de Serviços Técnicos (CGAL/DTEC).
A gestão deste manual está sob a responsabilidade da CGAL/DTEC que prestará auxílio ao público alvo leitor em caso de dúvidas e/ou sugestões quanto à aplicação deste manual, que devem ser submetidas ao Departamento responsável.
A publicação e atualização das versões na plataforma oficial da SDA para acesso pelo público alvo será de responsabilidade da Secretaria representada pelo DTEC.
¶ 3. Objetivo e Introdução
¶ 3.1 Objetivo
Este manual tem por objetivo estabelecer os métodos de análise físico-química, microscópica e microbiológica dos produtos destinados à alimentação animal, no âmbito da Rede Nacional de Laboratórios Agropecuários do Sistema Unificado de Atenção à Sanidade Agropecuária, consolidando o apoio laboratorial às ações de fiscalização do Ministério da Agricultura e Pecuária. Este manual também tem por objetivo contribuir para que a sociedade disponha de métodos analíticos adequados para o controle e a garantia da qualidade dos alimentos para animais consumidos no Brasil.
¶ 3.2 Introdução
Os métodos do presente manual são de aplicação compulsória pelos laboratórios oficiais do MAPA e pelos laboratórios credenciados integrantes da Rede Nacional de Laboratórios Agropecuários, substituindo as metodologias previamente publicadas em atos regulatórios como a Portaria n° 108/1991 (Métodos analíticos para controle de alimentos para uso animal) e a Instrução Normativa nº 69/2003 (Metodologia para detecção de subprodutos de origem animal em misturas de ingredientes para alimentação de ruminantes por microscopia).
Os métodos deste manual devem ter seu desempenho previamente verificado por seus executores em conformidade com a literatura técnica especializada, como os manuais do MAPA (Brasil, 2015) e Inmetro (Brasil, 2020) e demais publicações correlatas.
Em sua rotina analítica, cada laboratório deve monitorar a garantia da validade de seus resultados e atender os critérios de precisão e exatidão, quando constarem dos métodos indicados ou descritos neste manual.
Quando for solicitado pelo cliente, os resultados analíticos devem ser expressos com a incerteza de medição expandida (U), multiplicando-se a incerteza padrão combinada pelo coeficiente de abrangência (k) correspondente, com probabilidade de abrangência de aproximadamente 95%. Nesses casos, os resultados (R) de cada mensurando são informados como resultado ± incerteza de medição expandida, ambos expressos na unidade de grandeza do mensurando, com o mesmo número de casas decimais, da seguinte forma:
Mensurando: R ± U, k = x
Exemplos:
Umidade e voláteis: 140,1 ± 0,1 g/kg, k = 2
Extrato etéreo: 100,6 ± 0,5 g/kg, k = 2
Os cálculos dos resultados e da incerteza de medição, se realizados em planilhas eletrônicas, devem considerar todos os algarismos significativos, sendo arredondados no resultado. Quando os resultados forem calculados sem uso dessas planilhas, o arredondamento seguirá a norma ABNT NBR 5891. Caso seja utilizada outra regra de arredondamento, a mesma deverá ser informada no relatório de ensaio.
Produtos destinados à alimentação animal abrangem de matérias-primas a diversos ingredientes de origem animal, vegetal e mineral para fabricação de alimentos para animais, bem como uma ampla gama de alimentos prontos para o consumo de animais de estimação e de produção, entre outros. Diante dessa diversidade de matrizes, pode ser necessária a extensão de escopo de aplicação de determinados métodos de referência que originalmente não as contemplem. Nesses casos, quando uma nova matriz precisar ser incluída no escopo de aplicação de um método, as adaptações para sua utilização em produtos destinados à alimentação animal, bem como a inclusão de novos analitos, devem ser submetidas à validação analítica (se não houver método oficial para aquela combinação analito/matriz) e ser claramente descritas em procedimento operacional padrão do laboratório ou documento equivalente de seu sistema de gestão.
Para fins de aplicação dos métodos deste manual, todos os reagentes devem ser de grau analítico, exceto quando especificado. Toda a água utilizada nos procedimentos é destilada/deionizada ou ultrapura, exceto quando especificada.
¶ Bibliografia
Associação Brasileira de Normas Técnicas. Norma Brasileira ABNT NBR 5891. Regras de arredondamento na numeração decimal. Rio de Janeiro: ABNT, 2014. ISBN 978-85-07-05281-4.
Brasil. Ministério da Agricultura e Pecuária. Manual de garantia da qualidade analítica: áreas de identidade e qualidade de alimentos e de insumos/Ministério da Agricultura, Pecuária e Abastecimento. Secretaria de Defesa Agropecuária – Brasília: MAPA/ACS, 2015. 51 p. ISBN 978-85-7991-093-7.
Brasil. Ministério do Desenvolvimento, Indústria, Comércio e Serviços. Instituto Nacional de Metrologia, Qualidade e Tecnologia - Inmetro. Coordenação Geral de Acreditação. Orientação sobre validação de métodos analíticos. Documento de caráter orientativo DOQ-CGCRE-008 Revisão 09 – junho/2020.
¶ 4. Procedimentos
¶ A. Preparo de amostras
Todos os instrumentos, superfícies e recipientes destinados a receber e preparar as amostras para análise devem estar limpos e secos. Caso a análise não se inicie logo após o recebimento, armazenar as amostras de forma que suas características originais não sejam alteradas, protegendo-as da luz solar direta, ar e umidade.
Amostras líquidas ou pastosas devem ser transferidas para frascos com tampas (ambos identificados), que devem ser agitados antes da retirada das alíquotas necessárias às análises. Materiais viscosos podem ser aquecidos à temperatura máxima de 50 °C, pelo tempo mínimo necessário à redução de sua viscosidade, a fim de facilitar sua homogeneização.
Caso necessário, pode ser utilizado um processador de alimentos para homogeneização de amostras pastosas. Transferir a amostra juntamente com qualquer líquido para o recipiente do equipamento e processar por 30 segundos, raspando as laterais do recipiente com espátula e reincorporando esse material à porção principal da amostra, assim como qualquer material preso às lâminas. Processar por 30 segundos adicionais. Repetir este processo por duas vezes. Caso sejam utilizados equipamentos que realizam a raspagem simultaneamente ao processamento, pode ser feita uma única etapa de aproximadamente um minuto.
Amostras sólidas devem ser moídas e acondicionadas em frascos com boca larga e tampas para fechamento hermético ou em sacos plásticos (preferencialmente com fechamento), devidamente identificados. Após o preparo, as amostras devem ser mantidas sob proteção da luz, ar e umidade. Amostras heterogêneas poderão ser quarteadas de forma manual ou utilizando-se um quarteador adequado, até que se obtenha uma subamostra apropriada para a moagem, em quantidade suficiente às análises (no mínimo 100 g).
Instruções específicas de preparo das amostras, quando requeridas e descritas nos métodos analíticos, também devem ser observadas.
¶ A.1 Pré-secagem
Amostras sólidas com menos de 850 g/kg de matéria seca (umidade superior a 150 g/kg), destinadas às análises físico-químicas, deverão ser submetidas a uma etapa de pré-secagem (antes da moagem), para minimizar a potencial alteração do seu teor de umidade durante sua moagem e armazenamento. Nesses casos, os resultados das análises deverão ser expressos com a observação “amostra submetida à pré-secagem” e serão multiplicados por um fator de correção (Fc), calculado de acordo com a equação abaixo:
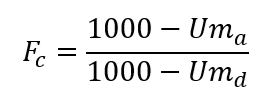
onde Uma = umidade (em g/kg) da amostra antes da moagem (determinada de acordo com o item B.10.1.4 deste manual); Umd = umidade (em g/kg) da amostra após a moagem (determinada de acordo com o item B.10.1.4 deste manual).
A pré-secagem deve ser feita a uma temperatura estável e inferior a 60 ºC (item B.10.1.5), de forma a evitar alterações da composição química da amostra durante o procedimento. Em seguida, a amostra deve ser mantida à temperatura ambiente por quinze minutos para em seguida ser moída e analisada.
¶ A.2 Moagem
A moagem das amostras sólidas deve ser feita rapidamente em moinho com refrigeração ou ultra centrífugo, evitando alterações de sua composição por ação do calor e reações de oxidação. Se necessário, pode-se fragmentar as amostras antes da moagem, com o auxílio de utensílios como tesouras, bisturis, martelos, chaves de fenda, espátulas, gral e pistilo.
Iniciar a moagem com a peneira de abertura maior, para em seguida ser feita com as de menor abertura. Exceto quando especificado, a moagem das amostras deve ser feita até que as partículas tenham dimensões de no máximo 1,0 mm. A cada moagem, as partículas remanescentes nas partes removíveis do moinho devem ser retiradas com auxílio de pincel e incorporadas à amostra. O moinho deve ser limpo após o processamento de cada amostra, a fim de evitar contaminação cruzada. Durante o uso, o limite de aquecimento do moinho não deve ser superado, respeitando os intervalos de tempo necessários ao processamento de muitas amostras.
Amostras com granulometria inferior a 1,0 mm podem ser dispensadas da etapa de moagem, desde que submetidas ao quarteamento.
Amostras destinadas à microscopia de rações não devem ser moídas.
Métodos analíticos específicos, como os que incluem etapas de filtração e são executados em sistemas automatizados, podem requerer moagem das amostras com peneiras de aberturas específicas. Nesses casos, seguir as recomendações do fabricante do instrumento, ou as especificadas no método.
¶ Bibliografia
- Brasil. Ministério da Agricultura, Pecuária e Abastecimento. Instrução Normativa nº 15 de 26 de maio de 2009. Regulamenta o registro dos estabelecimentos e dos produtos destinados à alimentação animal. Brasília: Diário Oficial da União (seção 1), 28 de maio de 2009.
- Compêndio Brasileiro de Alimentação Animal. 2023. Segurança em laboratórios e gerenciamento de riscos químicos. Apêndice A. Amostragem e preparo de amostras. São Paulo: Sindicato Nacional da Indústria da Alimentação Animal (2023).
- FAO. 2011. Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14. Rome.
- ISO 6498:2012(E). Animal feeding stuffs — Guidelines for sample preparation.
¶ B. Métodos físico-químicos
¶ B.1 Extrato etéreo
¶ B.1.1 Princípio
Este método determina gravimetricamente o teor de gordura total dos alimentos para animais. A gordura é extraída da amostra com éter etílico e/ou éter de petróleo. O solvente de extração é removido por destilação e secagem e o resíduo (extrato etéreo) é determinado por pesagem.
Amostras que em sua composição contenham produtos lácteos, leveduras, proteínas concentradas ou isoladas de glúten, soja ou batata, amostras de produtos submetidos a processamento térmico (extrusão) e amostras de sais cálcicos de ácidos graxos devem ser submetidas à hidrólise com ácido clorídrico, sob aquecimento, antes da extração da gordura. Outros produtos podem também ser submetidos à hidrólise ácida, desde que haja indicação técnica para o procedimento.
O método descrito neste manual se aplica a instrumentos automatizados para extração da gordura da amostra, podendo ser adaptado em função de características específicas de cada instrumento, ou por recomendação de seu fabricante. Alternativamente, outros métodos podem ser empregados, como a norma ISO 6492 ou os métodos AOAC 2003.06-2006 e AOAC 954.02-1977.
¶ B.1.2 Campo de aplicação
Este método se aplica a produtos destinados à alimentação animal, com exceção de sementes oleaginosas e seus resíduos.
¶ B.1.3 Materiais e equipamentos
- Algodão desengordurado
- Balança analítica com resolução mínima de 0,0001 g
- Balão volumétrico (100 mL)
- Béqueres ou erlenmeyers
- Cartuchos de celulose para extração
- Dessecador com placa de metal ou de porcelana espessa perfurada e agente dessecante (cloreto de cálcio anidro ou sílica gel)
- Estufa isotérmica (± 2 °C) de aquecimento elétrico, com regulação rápida da temperatura e convecção de ar
- Determinador de gordura com aquecimento elétrico
- Hidrolisador automático (opcional)
- Papel de filtração
- Papel de pesagem
- Pedras para controle da ebulição
- Pinça tenaz
- Placa aquecedora
- Provetas de 100 mL, 500 mL e 1000 mL
- Tubos de extração (“reboilers”)
- Vidro de relógio
¶ B.1.4 Reagentes e soluções
- Acetona (CH3(CO)CH3, CAS 67-64-1).
- Éter etílico (C4H10O, CAS 60-29-7) ou éter de petróleo.
- Solução de ácido clorídrico 4 mol/L: sob exaustão, diluir 333 mL de ácido clorídrico concentrado (HCl, CAS 7647-01-0, 12 mol/L) com água em uma proveta, completando volume para 1000 mL. Corrigir a concentração em função da densidade do ácido concentrado, caso seja necessário.
Nota: equipamentos de hidrólise automatizada podem requerer solução de ácido clorídrico em concentração diferente da usada neste método. Nesse caso, seguir as recomendações do fornecedor do instrumento.
- Solução de nitrato de prata 0,1%: dissolver 100 mg de nitrato de prata (AgNO3, CAS 7761-88-8) em água e transferir para um balão volumétrico de 100 mL. Completar volume com água. Homogeneizar e armazenar em frasco âmbar.
¶ B.1.5 Procedimento de análise
- Antes do uso, os tubos “reboilers” devem ser desengordurados, lavados, secos e tarados, para utilização a cada nova extração.
- Secar os tubos em estufa a 103 ºC ± 2 ºC por no mínimo uma hora. Retirar e esfriar em dessecador.
- Adicionar três a quatro pedras para controle da ebulição a cada tubo. Pesar e identificar.
¶ B.1.5.1 Hidrólise ácida automatizada
- Pesar de 2,00 a 5,00 g de amostra sobre o papel de pesagem.
- Acondicionar no béquer de hidrólise do equipamento.
- Adicionar papel de filtragem e fechar o compartimento.
- Verificar se o nível dos reservatórios de água e de ácido são suficientes e se o reservatório de descarte de resíduos está vazio.
- Iniciar a programação de hidrólise e lavagem, conforme recomendações do fabricante.
- A eficácia da lavagem pode ser avaliada recolhendo-se algumas gotas do filtrado em vidro de relógio e adicionando-se duas a três gotas da solução de nitrato de prata 0,1% (o resíduo da lavagem fica esbranquiçado na presença de cloreto). Outros testes poderão ser adotados, como a reação em papel de tornassol.
- Finalizada a hidrólise, retirar a amostra do equipamento e acondicionar em béquer. Levar à estufa para secagem por uma hora a 103 ºC ± 2 ºC.
- Esfriar em dessecador para posterior extração.
¶ B.1.5.2 Hidrólise ácida “manual”
- Pesar de 2,00 a 5,00 g de amostra homogeneizada em béquer de forma alta ou erlenmeyer e adicionar cerca de 50 mL de ácido clorídrico 4 mol/L.
- Homogeneizar com bastão de vidro e tampar com vidro de relógio.
- Aquecer em placa aquecedora por uma hora, agitando a cada dez minutos.
- Adicionar volumes de 150 mL de água quente, transferindo para recipiente de filtragem.
- Filtrar, desprezando o filtrado e lavando com água ultrapura, sem haver perda da amostra.
- Fazer o teste de cloreto, recolhendo algumas gotas do filtrado em vidro de relógio e adicionando nitrato de prata 0,1% ou papel de tornassol. O filtrado deve se encontrar livre de cloreto.
- Limpar o béquer e o bastão com algodão embebido em acetona e acondicionar o algodão junto com o filtro que contém o hidrolisado.
- Acondicionar o papel de filtragem em béquer e levar à estufa para secagem durante uma hora a 103 ºC ± 2 ºC.
¶ B.1.5.3 Extração etérea
- Operar o instrumento de extração de acordo com as recomendações do fabricante. Quando aplicável, ajustar a temperatura do banho de refrigeração do equipamento a 5 °C antes da extração.
- Adicionar o papel filtro ou o algodão com a amostra ao cartucho de celulose (se necessário, limpar o béquer com algodão desengordurado embebido em solvente de extração e acrescentar esse algodão ao cartucho de extração com a amostra).
- Adicionar os cartuchos aos tubos de extração juntamente com os suportes metálicos.
- Adicionar uma quantidade de solvente que seja suficiente para cobrir a parte do cartucho extrator na qual está alocada a amostra (em geral, utilizam-se volumes de 70 mL a 140 mL).
- Encaixar os tubos no equipamento.
- Selecionar a programação adequada. Processos de extração automatizada, em geral, duram de duas a quatro horas, incluindo etapas de imersão da amostra no solvente de extração e lavagem por refluxo do solvente. Se necessário, adicionar ao programa uma etapa de secagem ou realizar a mesma em placa de aquecimento (em banho maria), para evaporação do solvente remanescente.
- Secar os tubos em estufa a 103 ºC ± 2 ºC, por uma hora.
- Esfriar em dessecador e pesar.
- Repetir a secagem nas mesmas condições até que se tenha duas pesagens consecutivas com diferença menor que 0,1% entre elas.
¶ B.1.6 Cálculos e expressão dos resultados
O resultado deve ser expresso em “g/kg”, com uma casa decimal. Calcular o resultado conforme a equação abaixo, para cada replicata:
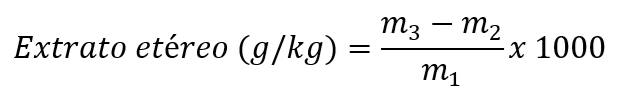
onde m1 = massa da amostra (g); m2 = massa do tubo ou balão (g); m3 = massa do tubo ou balão + extrato etéreo (g).
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 954.02-1977, Fat (crude) or ether extract in pet food. Gravimetric method.
- AOAC International. Official Methods of AOAC International. AOAC 2003.06-2006, Crude fat in feeds, cereal grains, and forages.
- ISO 6492:1999 Animal feeding stuffs — Determination of fat content.
¶ B.2 Fibra bruta
¶ B.2.1 Princípio
A fibra bruta é determinada gravimetricamente como o resíduo insolúvel obtido após duas etapas de hidrólise em meio aquoso, sob ebulição: uma ácida (com ácido sulfúrico), outra alcalina (com hidróxido de potássio ou hidróxido de sódio). Essas etapas de digestão quebram ligações de proteínas, lipídios e carboidratos, produzindo espécies químicas de estruturas menores, solúveis em meio aquoso. Após incineração, a massa do resíduo insolúvel, composto principalmente por celulose e lignina insolúvel em meio alcalino, corresponde à fibra bruta, quantificada por diferença da massa inicial da amostra.
Amostras com teores de gordura superior a 8% e de carbonato superior a 5% devem ser desengorduradas e neutralizadas antes da digestão.
O método descrito neste manual se aplica a instrumentos automatizados para determinação de fibra bruta com digestão das amostras em bolsas de filtro, podendo ser adaptado em função de características específicas de cada instrumento. Alternativamente, outros métodos podem ser empregados, como as normas ISO 5498 e ISO 6865 ou os métodos AOAC 962.09-1971(2010) e 2021.018 do Compêndio Brasileiro de Alimentação Animal.
¶ B.2.2 Campo de aplicação
Este método se aplica a ingredientes para rações, rações e concentrados, destinados à alimentação animal.
¶ B.2.3 Materiais e equipamentos
- Antiespumante (simeticona ou silicone)
- Balança analítica com resolução mínima de 0,0001 g
- Béqueres
- Bolsas de filtro (“fibre bags” ou “filter bags”)
Nota: a peneira utilizada na moagem da amostra deve ter abertura compatível com as especificações das bolsas de filtro, de forma a não propiciar perda por vazamento durante o processo analítico.
- Cadinhos de porcelana
- Conjunto digestor automatizado para análise de fibra com carrossel e espaçadores de vidro para acomodar as bolsas de filtro
- Dessecador com cloreto de cálcio ou sílica gel anidros
- Estufa de secagem
- Forno mufla
¶ B.2.4 Reagentes e soluções
- Éter de petróleo ou éter etílico (C4H10O, CAS 60-29-7)
- Solução de ácido clorídrico 0,5 mol/L: dispensar 21 mL de ácido clorídrico concentrado (HCl, CAS 7647-01-0) em um balão volumétrico de 500 mL, contendo cerca de 200 mL de água. Completar o volume com água e homogeneizar.
- Solução de ácido sulfúrico 0,127 mol/L: medir 7,0 mL de ácido sulfúrico concentrado (H2SO4, CAS 7664-93-9). Transferir para béquer de 1000 mL contendo aproximadamente 500 mL de água e aguardar esfriar. Transferir para balão volumétrico de 1000 mL, avolumar com água e homogeneizar. Adicionar duas gotas de antiespumante. Armazenar à temperatura ambiente.
- Solução de hidróxido de potássio 0,230 mol/L: pesar, em béquer, 15,20 g de hidróxido de potássio (KOH, CAS 1310-58-3). Se necessário, corrigir em função da pureza do reagente. Solubilizar com água e aguardar o resfriamento. Transferir quantitativamente para balão de 1000 mL, avolumar com água e homogeneizar. Adicionar duas gotas de antiespumante. Armazenar à temperatura ambiente.
- Solução de hidróxido de sódio 0,313 mol/L: pesar, em béquer, 12,50 g de hidróxido de sódio (NaOH, CAS 1310-73-2). Solubilizar com água e aguardar esfriar. Transferir quantitativamente para balão de 1000 mL, avolumar com água e homogeneizar. Adicionar duas gotas de antiespumante. Armazenar à temperatura ambiente.
¶ B.2.5 Procedimento de análise
¶ B.2.5.1 Determinação do branco
Toda vez que um lote de bolsas de filtro for aberto, analisar, no mínimo em duplicata, as cinzas do material isento de amostra, conforme procedimento a seguir:
- Tarar os cadinhos levando-os ao forno mufla por 30 minutos a 600 ºC.
- Esfriar os cadinhos em dessecador até temperatura ambiente.
- Pesar os cadinhos (massa m6).
- Tarar as bolsas de filtro secando-as em estufa a 105 ºC por uma hora.
- Esfriar as bolsas de filtro em dessecador até temperatura ambiente.
- Pesar as bolsas de filtro (esse valor não será utilizado nos cálculos) e colocá-las nos cadinhos identificados.
- Levar os conjuntos cadinho/bolsas de filtro ao forno mufla por no mínimo quatro horas a 600 ºC, até calcinação completa.
- Retirar os cadinhos com as cinzas da mufla quando a temperatura for superior a 105 °C, transferir para dessecador e aguardar resfriamento à temperatura ambiente.
- Pesar (massa m7).
- Calcular a média das replicatas.
¶ B.2.5.2 Pesagem das bolsas de filtro
- Tarar as bolsas de filtro, secando-as em estufa a 105 ºC por uma hora.
- Esfriar as bolsas de filtro em dessecador até temperatura ambiente.
- Pesar as bolsas de filtro (massa m1) e colocá-las nos espaçadores de vidro, atentando para o número de identificação de cada espaçador.
Nota: se as bolsas de filtro forem mantidas em ambiente anidro, esta etapa pode ser suprimida.
¶ B.2.5.3 Análise das amostras
- Pesar 1,00 g de amostra (massa m2) diretamente no conjunto bolsa de filtro/espaçador.
- Secar as amostras em estufa a 105 ºC por uma hora.
- Amostras com teor de gordura superior a 8% devem ser previamente desengorduradas com três porções sucessivas de éter de petróleo ou éter etílico, em volume suficiente para cobrir as amostras contidas nas bolsas de filtro. Substituir o solvente quando ficar visivelmente saturado. Manter as amostras sob exaustão até a evaporação do solvente.
- Amostras que em sua composição contenham teor de carbonato superior a 5% devem ser lavadas com três porções sucessivas de 30 mL (cada porção) de ácido clorídrico 0,5 mol/L para evitar a neutralização parcial do ácido sulfúrico durante a hidrólise ácida, lavando-se uma vez com 30 mL de água, antes da digestão.
- Inserir os conjuntos bolsas de filtro/espaçador/amostra no carrossel e alocá-lo no instrumento, iniciando o procedimento de digestão ácida (com ácido sulfúrico 0,127 mol/L) e básica (com hidróxido de potássio 0,230 mol/L ou hidróxido de sódio 0,313 mol/L), seguindo instruções do fabricante para operação do instrumento.
- Após a digestão, retirar o carrossel do equipamento.
- Retirar os espaçadores das bolsas de filtro, lavando-os cuidadosamente com água.
- Drenar o excesso de água das bolsas de filtro e colocá-las em cadinhos identificados.
- Levar os conjuntos cadinho/bolsas de filtro/amostra à estufa a 105 ºC por no mínimo quatro horas.
- Esfriar os conjuntos cadinho/bolsas de filtro/amostra em dessecador até temperatura ambiente.
- Pesar os conjuntos cadinho/bolsas de filtro/amostra (massa m3).
- Levar os conjuntos cadinho/bolsas de filtro/amostra ao forno mufla por no mínimo quatro horas a 600 ºC até calcinação completa.
- Retirar os conjuntos cadinho/cinzas da mufla em temperatura superior a 105 °C, transferir para dessecador e aguardar esfriar até a temperatura ambiente.
- Pesar os conjuntos cadinho/cinzas (massa m4).
¶ B.2.6 Cálculo e expressão dos resultados
Para cálculo do branco (m5), utilizar a equação:
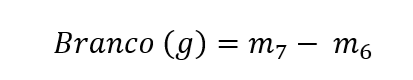
onde m6 = massa do cadinho seco (g); m7 = massa do cadinho seco + cinzas (g).
Para cálculo da fibra bruta (g/kg), utilizar a equação:
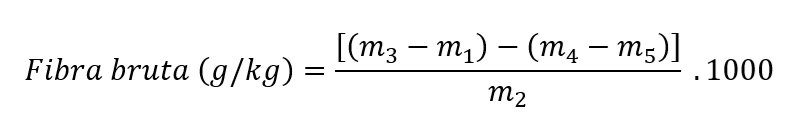
onde m1 = massa da bolsa de filtro seca (g); m2 = massa da amostra (g); m3 = massa do cadinho após digestão (g); m4 = massa do cadinho + cinzas (g); m5 = massa do branco da bolsa de filtro (g).
Expressar o resultado com uma casa decimal.
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 962.09-1971(2010), Fiber (crude) in animal feed and pet food.
- Compêndio Brasileiro de Alimentação Animal. 2023. Método 2021.018. Determinação de fibra bruta por hidrólise ácida e hidrólise básica. São Paulo: Sindicato Nacional da Indústria da Alimentação Animal (2023).
- ISO 5498:1981 Agricultural food products — Determination of crude fibre content — General method.
- ISO 6865:2000 Animal feeding stuffs — Determination of crude fibre content — Method with intermediate filtration.
¶ B.3 Fibra em detergente ácido
¶ B.3.1 Princípio
A fibra em detergente ácido é a fração do alimento que inclui celulose e lignina como componentes principais. É caracterizada como o resíduo insolúvel obtido após digestão da amostra, sob ebulição, em uma solução ácida constituída de ácido sulfúrico e brometo de cetiltrimetilamônio (CTAB). O processo de digestão quebra ligações de proteínas, lipídios e carboidratos, tornando-os solúveis. O resíduo resultante do processo é quantificado por gravimetria, descontando-se as cinzas.
Amostras com teor de gordura superior a 8% e teor de carbonatos superior a 5% (expresso como carbonato de sódio) devem ser desengorduradas e neutralizadas antes da digestão.
O método descrito neste manual se aplica a instrumentos automatizados para determinação de fibra em detergente ácido com digestão das amostras em bolsas de filtro, podendo ser adaptado em função de características específicas de cada instrumento. Alternativamente, outros métodos podem ser empregados, como a norma ISO 13906 ou o método AOAC 973.18-1977.
¶ B.3.2 Campo de aplicação
Este método se aplica a produtos destinados à alimentação animal.
¶ B.3.3 Materiais e equipamentos
- Antiespumante (simeticona ou silicone)
- Balança analítica com resolução mínima de 0,0001 g
- Béqueres
- Bolsas de filtro (“fibre bags” ou “filter bags”)
Nota: a peneira utilizada na moagem da amostra deve ter abertura compatível com as especificações das bolsas de filtro, de forma a não propiciar perda por vazamento durante o processo analítico.
- Cadinhos de porcelana
- Conjunto digestor automatizado para análise de fibra com carrossel e espaçadores de vidro para acomodar as bolsas de filtro
- Dessecador com cloreto de cálcio ou sílica gel anidros
- Estufa de secagem
- Forno mufla
¶ B.3.4 Reagentes e soluções
- Detergente ácido: brometo de cetiltrimetilamônio (CTAB, C19H42BrN, CAS 57-09-0), na concentração de 20 g/L de solução de ácido sulfúrico 0,5 mol/L (± 0,01 mol/L), previamente padronizada.
- Éter de petróleo ou éter etílico (C4H10O, CAS 60-29-7)
- Solução de ácido clorídrico 0,5 mol/L: dispensar 21 mL de ácido clorídrico concentrado (HCl, CAS 7647-01-0) em um balão volumétrico de 500 mL, contendo cerca de 200 mL de água. Completar o volume com água e homogeneizar.
¶ B.3.5 Procedimento de análise
¶ B.3.5.1 Determinação do branco
Toda vez que um lote de bolsas de filtro for aberto, analisar, no mínimo em duplicata, as cinzas do material isento de amostra, conforme procedimento a seguir:
- Tarar os cadinhos levando-os ao forno mufla por 30 minutos a 550 ºC. Cadinhos utilizados em rotina (que não sejam de primeiro uso) e que não perdem massa efetiva no processo de calcinação poderão apenas ser secos por no mínimo uma hora em estufa à temperatura de 105 ºC.
- Esfriar os cadinhos em dessecador até temperatura ambiente.
- Pesar os cadinhos (massa m6).
- Tarar as bolsas de filtro secando-as em estufa a 105 ºC por uma hora (dispensar esta etapa para bolsas de filtro guardadas em ambiente anidro).
- Esfriar as bolsas de filtro em dessecador até temperatura ambiente e colocá-las nos cadinhos identificados.
- Levar os conjuntos cadinho/bolsas de filtro ao forno mufla por no mínimo quatro horas a 550 ºC, até calcinação completa.
- Retirar os cadinhos com as cinzas da mufla quando a temperatura for superior a 105 °C, transferir para dessecador e aguardar resfriamento à temperatura ambiente.
- Pesar (massa m7).
- Calcular a média das replicatas.
¶ B.3.5.2 Pesagem das bolsas de filtro
- Tarar as bolsas de filtro, secando-as em estufa a 105 ºC por uma hora.
- Esfriar as bolsas de filtro em dessecador até temperatura ambiente.
- Pesar as bolsas de filtro (massa m1) e colocá-las nos espaçadores de vidro, atentando para o número de identificação de cada espaçador.
Nota: se as bolsas de filtro forem mantidas em ambiente anidro, esta etapa pode ser suprimida.
¶ B.3.5.3 Digestão das amostras
- Pesar 1,00 g de amostra (massa m2) diretamente no conjunto bolsa de filtro/espaçador.
- Após o procedimento de pesagem, amostras úmidas ou semiúmidas deverão ser secas em estufa a 105 ºC, por tempo suficiente para a remoção do excesso de umidade.
- Amostras com teor de gordura superior a 8% devem ser previamente desengorduradas com três porções sucessivas de éter de petróleo ou éter etílico, em volume suficiente para cobrir as amostras contidas nas bolsas de filtro. Substituir o solvente quando ficar visivelmente saturado. Manter as amostras sob exaustão até a evaporação do solvente.
- Amostras que em sua composição contenham teor de carbonatos superior a 5% devem ser lavadas com três porções sucessivas de 30 mL (cada porção) de ácido clorídrico 0,5 mol/L para evitar a neutralização parcial do ácido sulfúrico durante a digestão, lavando-se uma vez com 30 mL de água, removendo em seguida o excesso de umidade, em estufa à 105 ºC.
- Inserir os conjuntos bolsas de filtro/espaçador/amostra no carrossel e alocá-lo no instrumento, iniciando o procedimento de digestão em detergente ácido, seguindo instruções do fabricante para operação do instrumento.
- Após a digestão, retirar o carrossel do equipamento.
- Retirar os espaçadores das bolsas de filtro, lavando-os cuidadosamente com água.
- Drenar o excesso de água das bolsas de filtro e colocá-las em cadinhos identificados.
- Levar os conjuntos cadinho/bolsas de filtro/amostra à estufa a 105 ºC por no mínimo quatro horas.
- Esfriar os conjuntos cadinho/bolsas de filtro/amostra em dessecador até temperatura ambiente.
- Pesar os conjuntos cadinho/bolsas de filtro/amostra (massa m3).
- Levar os conjuntos cadinho/bolsas de filtro/amostra ao forno mufla por no mínimo quatro horas a 550 ºC até calcinação completa.
- Retirar os conjuntos cadinho/cinzas da mufla em temperatura superior a 105 °C, transferir para dessecador e aguardar esfriar até a temperatura ambiente.
- Pesar os conjuntos cadinho/cinzas (massa m4).
¶ B.3.6 Cálculo e expressão dos resultados
Para cálculo do branco (m5), utilizar a equação:
onde m6 = massa do cadinho seco (g); m7 = massa do cadinho seco + cinzas (g).
Para cálculo da fibra em detergente ácido (g/kg), utilizar a equação:
onde m1 = massa da bolsa de filtro seca (g); m2 = massa da amostra (g); m3 = massa do cadinho após digestão (g); m4 = massa do cadinho + cinzas (g); m5 = massa do branco da bolsa de filtro (g).
Expressar o resultado com uma casa decimal.
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 973.18-1977, Fiber (acid detergent) and lignin (H2SO4) in animal feed.
- ISO 13906:2008 Animal feeding stuffs — Determination of acid detergent fibre (ADF) and acid detergent lignin (ADL) contents.
¶ B.4 Lignina em detergente ácido
Determinar o teor de lignina em detergente ácido, utilizando o método descrito na norma ISO 13906 ou o método AOAC 973.18-1977, expressando o resultado obtido em “g/kg”, com uma casa decimal.
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 973.18-1977 Fiber (acid detergent) and lignin (H2SO4) in animal feed.
- ISO 13906:2008 Animal feeding stuffs — Determination of acid detergent fibre (ADF) and acid detergent lignin (ADL) contents.
¶ B.5 Fibra em detergente neutro
Determinar o teor de fibra em detergente neutro, utilizando o método descrito na norma ISO 16472 ou o método AOAC 2002.04-2005, expressando o resultado obtido em “g/kg”, com uma casa decimal.
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 2002.04-2005, Amylase-treated neutral detergent fiber in feeds.
- ISO 16472:2006 Animal feeding stuffs — Determination of amylase-treated neutral detergent fibre content (aNDF).
¶ B.6 Índice de peróxido
Utilizar o método descrito na norma ISO 3960 para determinação do índice de peróxido na porção gordurosa da amostra, expressando o resultado final em “meq de O2/kg de gordura”, com uma casa decimal.
Nota 1: a análise de índice de peróxido deve preferencialmente ser executada logo após recebimento da amostra no laboratório. Se for necessário o armazenamento da amostra antes da análise, mantê-la em local fresco ou refrigerado, sob proteção da luz.
Nota 2: para extração da porção gordurosa, não é necessária a moagem da amostra. A quantidade mínima de gordura a ser extraída é 0,5 g/réplica do ensaio. A extração da gordura pode ser feita de modo direto, com a utilização de solvente isento de peróxido (éter de petróleo ou éter etílico), seguida de filtração e eliminação do solvente em condições brandas. O ensaio deve ser executado em, no mínimo, duplicata.
¶ Bibliografia
- ISO 3960:2017 Animal and vegetable fats and oils — Determination of peroxide value — Iodometric (visual) endpoint determination.
¶ B.7 Macro e micronutrientes minerais
¶ B.7.1 Mineralização da amostra
A disponibilização dos analitos em uma solução (“abertura" ou mineralização da amostra) para análise de macro e micronutrientes minerais pelos métodos deste manual pode ser feita por simples dissolução da amostra (amostras de produtos de origem mineral) em meio ácido ou por prévia oxidação de sua matéria orgânica, seja por via seca (calcinação), seja por via úmida (digestão aberta ou fechada).
O branco de análise deve ser preparado simultaneamente ao processamento das amostras, com todos os reagentes em quantidades iguais às utilizadas nas amostras.
Quando houver instruções específicas para a mineralização das amostras nos métodos analíticos, as mesmas devem ser seguidas.
Nota: recomenda-se a desmineralização dos tubos de digestão, cadinhos e vidraria de diluição das amostras com ácido nítrico antes de sua reutilização.
¶ B.7.1.1 Digestão por via úmida aberta
Seguir o procedimento descrito no método AOAC 935.13-1951, adicionando 25 mL de ácido nítrico para massas de 1,00 g a 2,50 g de cada amostra em béqueres identificados. Cobrir cada béquer com vidro de relógio e transferir para chapa de aquecimento sob calor baixo. Diluir para balão de 100 mL, completando o volume com água. Diluições subsequentes com ácido clorídrico (0,1 mol/L a 0,5 mol/L) podem ser necessárias para inserir as amostras na faixa analítica.
¶ B.7.1.2 Digestão por micro-ondas (via úmida fechada)
A proporção entre a amostra e os reagentes de digestão deve ser definida com base na matriz em análise e nas especificações operacionais do forno de micro-ondas empregado no procedimento. Em geral, são utilizados como reagentes de digestão (isolados ou combinados) ácido nítrico, ácido clorídrico, ácido fluorídrico, ácido sulfúrico e/ou peróxido de hidrogênio e massas de 0,50 g a 2,00 g de amostra. Após a adição da amostra e dos reagentes aos tubos de digestão, seguir as instruções a seguir:
- Preparar um branco de reagentes a cada batelada de digestão.
- Deixar os tubos em repouso por no mínimo 30 minutos na capela de exaustão, para pré-digestão.
- Tampar os tubos e seguir as instruções para digestão por micro-ondas, selecionando o método instrumental com tempo, temperatura e potência de digestão apropriados, utilizando rampa de aquecimento.
- Após resfriamento, destampar os tubos, rinsando as tampas.
- Acondicionar os tubos no banho de ultrassom por 15 minutos. Verificar se a amostra foi completamente digerida e filtrar ou fazer nova digestão, se for necessário.
- Transferir o conteúdo dos tubos para balões volumétricos e avolumar com água.
- Fazer diluições apropriadas, levando em conta a faixa linear e os limites analíticos.
Nota: a digestão de amostras que contenham silicatos ou dióxido de titânio pode requerer a adição de ácido fluorídrico. Nesses casos, seguir as medidas de segurança apropriadas à sua manipulação e empregar materiais compatíveis com o seu uso no procedimento analítico.
¶ B.7.1.3 Via seca (calcinação)
- Pesar entre 2,00 g a 10,00 g de amostra em cadinho de porcelana, de acordo com o requerido pelo método específico.
- Calcinar em forno mufla, programando uma rampa de aquecimento até a temperatura de calcinação (550 ºC), a qual deve ser mantida por no mínimo 240 minutos. Alternativamente, a carbonização das amostras pode ser feita em placa aquecedora, antes da calcinação.
- Resfriar os cadinhos na mufla ou em dessecador.
- Solubilizar as cinzas obtidas com ácido clorídrico e/ou ácido nítrico, de acordo com as instruções do método específico.
- Aquecer em placa aquecedora para facilitar a solubilização. Se necessário, utilizar vidro de relógio, mantendo por 10 a 15 minutos após condensação dos vapores, acompanhando o processo com atenção para evitar perda de amostra.
- Filtrar, recolhendo o filtrado em balão volumétrico (100 mL, 200 mL, 250 mL ou 500 mL).
- Lavar o resíduo do cadinho e o papel de filtração com água por no mínimo três vezes. Avolumar com água e homogeneizar.
Nota: amostras de produtos de origem mineral não necessitam de calcinação, apenas a solubilização com ácido clorídrico e/ou ácido nítrico em chapa aquecedora.
¶ Bibliografia
AOAC International. Official Methods of AOAC International. AOAC 935.13-1951, Calcium in animal feed. Wet ash method.
AOAC International. Official Methods of AOAC International. AOAC 968.08-1969(1996), Minerals in animal feed and pet food. Atomic absorption spectrophotometric method.
¶ B.7.2 Cálcio (determinação por volumetria de oxirredução)
Utilizar o método descrito na norma ISO 6490-1, expressando o resultado como um número inteiro em “mg/kg” (se menor ou igual a 10000 mg/kg) ou em “g/kg”, com uma casa decimal (se maior que 10 g/kg). Alternativamente, pode ser empregado o método 2021.004 do Compêndio Brasileiro de Alimentação Animal.
¶ Bibliografia
- Compêndio Brasileiro de Alimentação Animal. 2023. Método 2021.004. Determinação de cálcio pelo método oxidimétrico. São Paulo: Sindicato Nacional da Indústria da Alimentação Animal (2023).
- ISO 6490-1:1985 Animal feeding stuffs — Determination of calcium content. Part 1: Titrimetric method.
¶ B.7.3 Flúor (determinação por potenciometria)
Determinar o teor de flúor na amostra de acordo com o método AOAC 975.08-1976, expressando o resultado obtido como um número inteiro em “mg/kg” (se menor ou igual a 10000 mg/kg) ou em “g/kg”, com uma casa decimal (se maior que 10 g/kg). Quando necessário expressar o resultado da relação fósforo/flúor (relação P/F), utilizar a equação do item B.7.4.1.
Alternativamente, determinar o flúor utilizando o método descrito no item B.7.3.1 deste manual.
¶ B.7.3.1 Flúor (determinação por potenciometria com extração assistida por ultrassom)
¶ Princípio
O flúor é determinado como íon fluoreto por potenciometria direta, utilizando um potenciômetro acoplado a um eletrodo íon-seletivo para fluoreto, cujo elemento sensível é constituído por um monocristal de fluoreto de lantânio. O eletrodo, ao entrar em contato com a solução em análise, gera uma diferença de potencial elétrico (mV) proporcional à atividade do íon fluoreto. Esse sinal é correlacionado à concentração do analito (em mg/L) por meio de uma curva de calibração preparada com soluções-padrão de flúor. A adição de solução tampão tem como finalidade ajustar o pH do ambiente químico, mantendo a força iônica constante, para liberar o íon fluoreto de complexos químicos e minimizar interferências, assegurando resposta seletiva e reprodutível do eletrodo.
¶ B.7.3.1.1 Campo de aplicação
Este método se aplica a amostras de suplementos minerais e ingredientes de origem mineral destinados à alimentação animal.
¶ B.7.3.1.2 Materiais e equipamentos
- Balões volumétricos
- Banho de ultrassom
- Béqueres
- Estufa para secagem
- Funis
- Micropipetas
- Papel de filtração
- Pipetas Pasteur
- Potenciômetro acoplado a eletrodo de referência e sensor íon seletivo para flúor
- Tubos cônicos de polipropileno (tipo falcon) com capacidade de 50 mL
¶ B.7.3.1.3 Reagentes e soluções
- Solução de ácido clorídrico 1 mol/L: em um balão volumétrico de 1 L, adicionar 500 mL de água e em seguida adicionar cuidadosamente 83,3 mL de ácido clorídrico (HCl, CAS 7647-01-0) 37% (v/v). Completar o volume com água e homogeneizar.
- Solução de acetato de sódio 3 mol/L: dissolver 408 g de acetato de sódio trihidratado (CH3COONa.3H2O, CAS 6131-90-4) em água e transferir para balão volumétrico de 1 L. Quando a solução atingir a temperatura ambiente, completar o volume com água e homogeneizar. Ajustar o pH a 7,0 com algumas gotas de ácido acético (CH3COOH, CAS 64-19-7). Opcionalmente, dissolver 246,1 g de acetato de sódio monohidratado (CH₃COONa.H₂O, CAS 41484‑91‑7) em água e transferir para balão volumétrico de 1 L. Quando a solução atingir a temperatura ambiente, completar o volume com água e homogeneizar. Ajustar o pH a 7,0 com algumas gotas de ácido acético.
- Solução de citrato de sódio 1,32 mol/L: dissolver 222 g de citrato de sódio dihidratado (Na3C6H5O7.2H2O, 6132-04-3) com cerca de 250 mL de água e transferir para balão volumétrico de 1 L. Adicionar 28 mL de ácido perclórico (HClO4, CAS 7601-90-3). Completar o volume com água e homogeneizar.
- Solução de cloreto de potássio 3 mol/L (para limpeza do eletrodo): pesar 5,6 g de cloreto de potássio (KCl, CAS 7447-40-7) e transferir para um balão volumétrico de 25 mL. Completar o volume com água e homogeneizar. Essa solução deve ser preparada no momento do uso e não deve ser armazenada.
- Solução-padrão estoque de flúor (500 mg/L): pesar com exatidão 1,105 g de fluoreto de sódio (NaF, CAS 7681-49-4), previamente seco em estufa por quatro horas a aproximadamente 100 °C) e transferir para balão volumétrico de 1 L. Avolumar com água, homogeneizar e transferir para frasco plástico com tampa. Opcionalmente, usar solução padrão de fluoreto de sódio (1000 mg/L).
- Solução-padrão de trabalho intermediária (100 mg/L): dispensar 20 mL da solução estoque de fluoreto de sódio (500 mg/L) em balão volumétrico de 100 mL. Completar o volume com água e homogeneizar. Opcionalmente, dispensar 10 mL da solução estoque de fluoreto de sódio (1000 mg/L) em balão volumétrico de 100 mL. Completar o volume com água e homogeneizar.
¶ B.7.3.1.4 Procedimento de análise
¶ B.7.3.1.4.1 Curva de calibração
- Em balões volumétricos de 50 mL, dispensar alíquotas das soluções intermediárias de 100 mg/L para preparar a curva de calibração (tabela 1).
- Adicionar, a cada balão, 20 mL de ácido clorídrico 1 mol/L, 12,5 mL de acetato de sódio (3 mol/L) e 12,5 mL de citrato de sódio (1,32 mol/L). Completar os volumes com água e homogeneizar.

¶ B.7.3.1.4.2 Controle de recuperação
- Preparar uma triplicata do ponto da curva de calibração com concentração de 1 mg/L.
- Quando disponíveis, amostras controle devem ser também analisadas.
¶ B.7.3.1.4.3 Extração assistida por ultrassom (UAE)
- Considerando o teor esperado de flúor na amostra, pesar em tubos cônicos tipo falcon: amostras com baixo teor de flúor (ex.: suplemento mineral orgânico ou ração): pesar até 0,5 g; amostras com alto teor de flúor (ex.: suplemento mineral): pesar no mínimo 0,1 g. A massa de amostra deve ser ajustada de forma a manter sua concentração de flúor dentro da faixa de trabalho da curva analítica.
- Adicionar 20 mL de ácido clorídrico 1 mol/L e agitar em vórtex por 30 segundos.
- Submeter a amostra a banho de ultrassom (60 Hz) por 20 minutos.
- Filtrar a solução utilizando papel de filtro, coletando o filtrado no próprio balão volumétrico de 50 mL.
- Adicionar 12,5 mL de acetato de sódio (3 mol/L).
- Adicionar 12,5 mL de citrato de sódio (1,32 mol/L).
- Completar o volume com água, homogeneizar e proceder à determinação do flúor.
¶ B.7.3.1.4.4 Determinação
- Conectar o eletrodo íon seletivo de fluoreto e o eletrodo de referência ao potenciômetro e ligar o equipamento.
- Condicionar adequadamente o sistema e imergir os eletrodos na solução sem adição de flúor (branco) por, no mínimo, 30 minutos para estabilização.
- Dispensar as soluções de calibração e das amostras em análise em béqueres previamente identificados.
- Para a realização das medições, mergulhar os eletrodos em cada uma das soluções.
- Após cada leitura, enxaguar cuidadosamente os eletrodos e a barra de agitação com água deionizada, secando-os com papel absorvente limpo, a fim de evitar contaminações cruzadas.
- Iniciar a determinação pela leitura do branco.
- Prosseguir com as leituras das soluções da curva de calibração em ordem crescente de concentração (0,1 a 10 mg/L), respeitando um intervalo de estabilização entre cada leitura.
- Construir a curva de calibração utilizando os logaritmos das concentrações dos padrões e os respectivos sinais analíticos obtidos. Em seguida, aplicar a equação de regressão gerada para interpolar os resultados das amostras. Antes disso, verificar se os sinais analíticos das amostras estão dentro da faixa linear da curva. Caso estejam fora, será necessário ajustar a quantidade de amostra ou realizar uma diluição apropriada e repetir o procedimento.
- Amostras que ultrapassem o limite superior da curva deverão ser diluídas em água e reanalisadas.
Nota 1: As condições da leitura potenciométrica das amostras (tempo de estabilização, temperatura e agitação constante da solução) devem ser as mesmas utilizadas para a curva de calibração.
Nota 2: O orifício do eletrodo deve permanecer aberto durante a leitura. Caso o nível de solução do eletrodo diminua, completar com cloreto de potássio (3 mol/L).
Nota 3: Se surgirem cristais dentro do eletrodo, aspirar seu conteúdo com auxílio de uma pipeta Pasteur e lavar seu interior com cloreto de potássio (3 mol/L), até a dissolução dos cristais. Completar com cloreto de potássio (3 mol/L). Alternativamente, se existentes, devem ser seguidas as orientações de limpeza com as soluções recomendadas pelo fornecedor do instrumento.
¶ B.7.3.1.4.5 Cálculos e expressão dos resultados
A concentração de flúor é determinada (em mg/kg) de acordo com a equação:
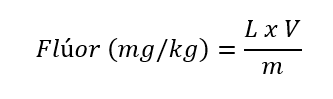
onde L = concentração (mg/L) calculada pela equação de regressão; V = volume do balão de diluição; m = massa da amostra (em g).
Os resultados obtidos devem ser expressos como um número inteiro em “mg/kg” (se menor ou igual a 10000 mg/kg) ou em “g/kg”, com uma casa decimal (se maior que 10 g/kg).
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 975.08-1976, Fluorine in animal feed, ion selective electrode method.
¶ B.7.4 Fósforo (determinação por espectrofotometria de absorção molecular)
Determinar o fósforo total utilizando o método descrito na norma ISO 6491:1998. Opcionalmente, utilizar o método descrito no manual FAO Animal Production and Health – Manual quality assurance for animal feed analysis laboratories, 2011 (página 132). Expressar o resultado como um número inteiro em “mg/kg” (se menor ou igual a 10000 mg/kg) ou em “g/kg”, com uma casa decimal (se maior que 10 g/kg).
¶ B.7.4.1 Relação fósforo/flúor (relação P/F)
Quando necessário, utilizar a equação abaixo para calcular a relação P/F, inserindo os resultados de cada determinação na mesma unidade de medida (mg/kg ou g/kg):

¶ Bibliografia
- FAO. 2011. Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14. Rome.
- ISO 6491:1998 Animal feeding stuffs — Determination of phosphorus content — Spectrometric method.
¶ B.7.5 Determinação de cálcio, cobalto, cobre, ferro, magnésio, manganês, potássio, sódio e zinco por espectrometria de absorção atômica com chama
Determinar o teor de cálcio, cobalto, cobre, ferro, magnésio, manganês, sódio e potássio e zinco de acordo com a norma ISO 6869 ou pelo método AOAC 968.08-1969(1996), expressando o resultado obtido para cada elemento como um número inteiro em “mg/kg” (se menor ou igual a 10000 mg/kg) ou em “g/kg”, com uma casa decimal (se maior que 10 g/kg).
Alternativamente, potássio e sódio podem ser determinados por espectrometria de emissão atômica com chama, utilizando-se o método descrito no item B.7.7 deste manual.
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 968.08-1969(1996), Minerals in animal feed and pet food. Atomic absorption spectrophotometric method.
- ISO 6869:2000 - Animal feeding stuffs — Determination of the contents of calcium, copper, iron, magnesium, manganese, potassium, sodium and zinc — Method using atomic absorption spectrometry.
¶ B.7.6 Determinação de cálcio, cobalto, cobre, cromo, ferro, fósforo, magnésio, manganês, potássio, selênio, sódio e zinco por espectrometria de emissão ótica por plasma indutivamente acoplado
¶ B.7.6.1 Princípio
A amostra é submetida à digestão ácida e diluída. A solução é introduzida na fonte de plasma de argônio (pureza mínima: 99,999%) indutivamente acoplado. Nesse processo, a solução é transformada em aerossol e transportada ao plasma gerado por uma fonte de argônio à alta temperatura por radiofrequência. Os elementos presentes na amostra são atomizados e ionizados, emitindo radiação em diferentes comprimentos de onda. As radiações emitidas são separadas por um sistema ótico e sua intensidade é medida pelo detector, que a correlaciona com a concentração do analito, definida em curvas de calibração.
¶ B.7.6.2 Campo de aplicação
Este método se aplica à determinação quantitativa de metais alcalinos (sódio e potássio), metais alcalinos terrosos (cálcio e magnésio), metais de transição (cobalto, cromo, cobre, ferro, manganês e zinco) e não metais (fósforo e selênio) em produtos destinados à alimentação animal.
¶ B.7.6.3 Materiais e equipamentos
- Balança analítica com resolução mínima de 0,0001 g
- Balões volumétricos
- Banho de ultrassom
- Espectrômetro de emissão ótica por plasma indutivamente acoplado (ICP-OES)
- Funis
- Papel de filtração (preferencialmente quantitativo)
- Micropipetas de volume ajustável e ponteiras
¶ B.7.6.4 Reagentes e soluções
- Ácido clorídrico (HCl, CAS 7647-01-0)
- Ácido nítrico (HNO3, CAS 7697-37-2)
- Peróxido de hidrogênio (H2O2, CAS 7722-84-1)
- Solução-padrão estoque (1000 mg/L) de cada elemento ou multielementar em meio ácido. Todas as soluções-padrão (estoques e soluções de trabalho) devem ser preferencialmente acondicionadas em frascos plásticos após seu preparo.
¶ B.7.6.5 Procedimento de análise
¶ B.7.6.5.1 Disponibilização dos analitos
Seguir um dos procedimentos descritos no item B.7.1 para disponibilização dos analitos.
¶ B.7.6.5.2 Curva de calibração
- A curva de calibração deve ser preparada com cinco diferentes concentrações de cada analito, assegurando que a concentração de ácido nítrico seja a mesma das soluções das amostras.
- Caso necessário, recomenda-se o preparo de curvas de calibração que abranjam distintas faixas de trabalho (curvas de baixa e de alta concentração), considerando a configuração ótica do ICP-OES (vista axial ou radial).
- Quando for utilizada calibração com padronização interna, a concentração do padrão interno adicionado às amostras e à curva de calibração deve ser a mesma.
¶ B.7.6.5.3 Condições instrumentais
A seleção dos comprimentos de onda utilizados para determinação dos analitos e dos padrões internos (quando aplicável) deve ser realizada com base na configuração instrumental definida em validação analítica, nas características espectrais dos elementos e potenciais interferentes e nos requisitos do método quanto à sua linearidade e sensibilidade. Na tabela 1, são propostos comprimentos de onda para determinação dos analitos.
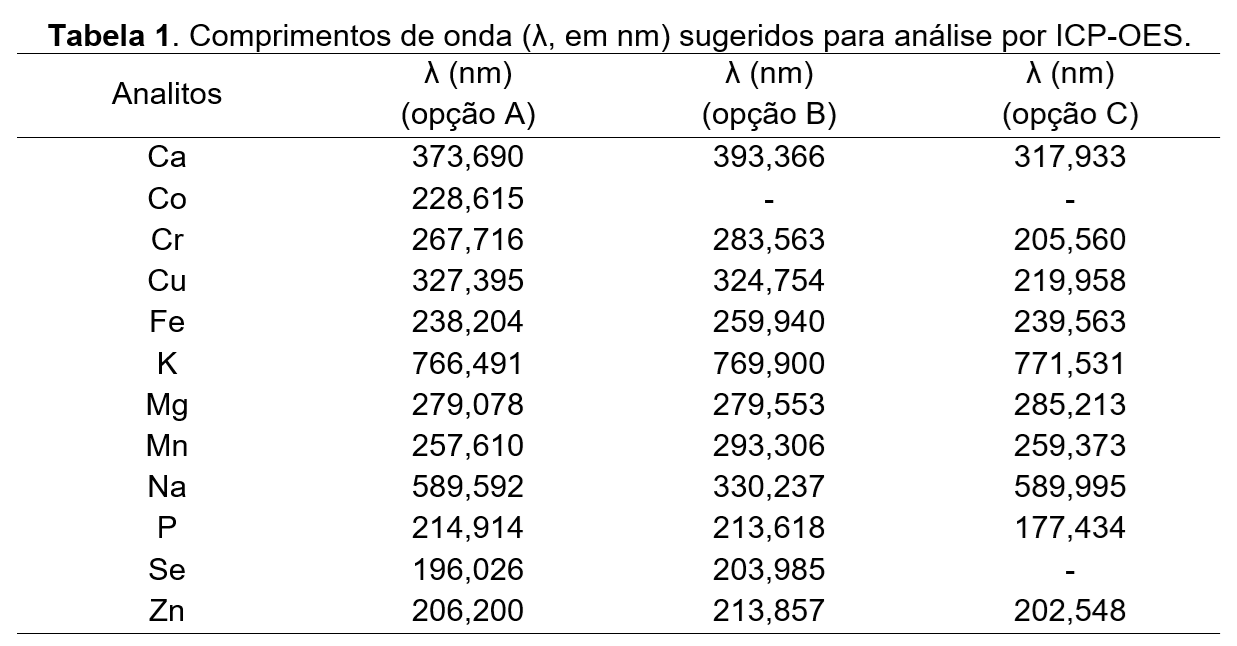
Ajustadas as condições operacionais, fazer a purga do instrumento e ignizar o plasma, aguardando o tempo de estabilização. Fazer o teste de calibração das linhas de emissão com solução multielementar adequada. Ao término das leituras de cada amostra, aspirar água ou ácido nítrico diluído (1% a 5%) por aproximadamente 10 segundos. Ao final da análise, passar água ou ácido nítrico diluído (1% a 5%) pelo capilar de amostragem por pelo menos cinco minutos.
¶ B.7.6.6 Cálculos e expressão dos resultados
Utilizando o método dos mínimos quadrados, interpolar na equação de regressão da curva de calibração de cada analito (concentrações dos padrões x sinais analíticos) o sinal analítico da amostra. Ajustar o resultado em função das diluições e massa de partida. Expressar o resultado de cada elemento com um número inteiro em “mg/kg” (se menor ou igual a 10000 mg/kg) ou em “g/kg”, com uma casa decimal (se maior que 10 g/kg). Aceitar as curvas cujos coeficientes de regressão (R2) sejam iguais ou superiores a 0,99.
¶ Bibliografia
- Compêndio Brasileiro de Alimentação Animal. 2023. Método 2021.041. Determinação de constituintes inorgânicos por espectrometria de emissão ótica por plasma acoplado indutivamente (ICP-OES). São Paulo: Sindicato Nacional da Indústria da Alimentação Animal (2023).
- European Committee for Standardization. European Standard EN 15621:2012. Animal feeding stuffs: Methods of sampling and analysis - Determination of calcium, sodium, phosphorus, magnesium, potassium, sulphur, iron, zinc, copper, manganese and cobalt after pressure digestion by ICP-AES.
¶ B.7.7 Determinação de sódio e potássio por espectrometria de emissão atômica com chama
¶ B.7.7.1 Princípio
A amostra é transformada em solução ácida de acordo com um dos procedimentos descritos no item B.7.1, na qual ficam disponíveis para quantificação os metais alcalinos.
A espectrometria de emissão atômica em chama (FAES) se baseia na medida da radiação eletromagnética emitida nas regiões visível e ultravioleta do espectro eletromagnético por átomos neutros ou ionizados excitados. Os analitos, ao receberem energia da chama, geram espécies excitadas que, ao retornarem para o estado fundamental, liberam parte da energia recebida na forma de radiação, em comprimentos de onda específicos.
¶ B.7.7.2 Campo de aplicação
Este método se aplica à determinação quantitativa de metais alcalinos (sódio e potássio) em produtos destinados à alimentação animal.
¶ B.7.7.3 Materiais e equipamentos
- Balança analítica com resolução mínima de 0,0001 g
- Balões volumétricos de 50 mL, 100 mL, 200 mL, 250 mL e/ou 500 mL
- Fotômetro de chama
- Funis
- Papel de filtração quantitativa
- Proveta graduada
¶ B.7.7.4 Reagentes e soluções
- Ácido nítrico (HNO3, CAS 7697-37-2)
- Cloreto de potássio (KCl, CAS 7447-40-7), padrão sólido ou em solução-padrão (1000 mg/L)
- Cloreto de sódio (NaCl, CAS 7647-14-15), padrão sólido ou em solução-padrão (1000 mg/L)
- Solução-padrão estoque (1000 mg/L) de cada elemento ou multielementar em meio ácido
- Solução-estoque de sódio e potássio 100 mg L-1 (preparada com padrões sólidos): secar os padrões (NaCl e KCl) em estufa a 110 °C por duas horas. Esfriar em dessecador. Pesar 0,254 g de NaCl e 0,1907 g de KCl e transferir para um balão de 1 L. Avolumar com água adicionando ácido nítrico até que sua concentração seja 2% e homogeneizar.
- Solução-estoque de sódio e potássio 100 mg L-1 (solução-padrão): transferir 10 mL de solução-padrão de sódio (1000 mg L-1) e 10 mL de solução-padrão de potássio (1000 mg L-1) para um balão volumétrico de 100 mL. Avolumar com água, adicionando ácido nítrico até que sua concentração seja 2%, e homogeneizar.
- Solução de ácido nítrico 1:4 (v/v): adicionar aproximadamente 500 mL de água a um balão de 1000 mL. Adicionar 200 mL de HNO3 cuidadosamente pela parede do balão. Avolumar com água e homogeneizar.
Nota: todas as soluções-padrão (estoques e soluções de trabalho) devem ser acondicionadas em frascos plásticos após seu preparo e mantidas sob refrigeração.
¶ B.7.7.5 Procedimento de análise
¶ B.7.7.5.1 Curva de calibração
- A curva de calibração deve ser preparada com diferentes concentrações de cada analito, fazendo diluições da solução-estoque 100 mg/L em balões volumétricos. Adicionar ácido nítrico concentrado de forma que sua concentração seja 2% em cada balão.
- Fazer um branco da curva de calibração com solução de ácido nítrico (2%).
- A calibração deve ser feita com, no mínimo, quatro pontos (2 mg/L, 5 mg/L, 10 mg/L e 20 mg/L), ou de acordo com as recomendações do fabricante do equipamento e a faixa linear requerida para determinação dos analitos nas amostras.
- Aceitar as curvas cujos coeficientes de regressão (R2) sejam iguais ou superiores a 0,99.
¶ B.7.7.5.2 Abertura da amostra e determinação
- Pesar entre 2,00 g e 4,00 g de amostra, seguindo um dos procedimentos descritos no item B.7.1 para disponibilização dos analitos.
- Se utilizada a mufla, a temperatura de calcinação deve ser 525 ºC, mantida por no mínimo 240 minutos. Solubilizar as cinzas obtidas com 15 mL de ácido nítrico 1:4 (v/v). Aquecer em placa aquecedora para facilitar a solubilização. Se necessário, utilizar vidro de relógio, mantendo por 10 a 15 minutos após condensação dos vapores, acompanhando o processo com atenção para evitar perda de amostra. Filtrar, recolhendo o filtrado em balão volumétrico (100 mL, 200 mL, 250 mL ou 500 mL). Lavar o resíduo do cadinho e o papel de filtração com água por no mínimo três vezes. Avolumar com água e homogeneizar.
- Ler a solução obtida diretamente no fotômetro de chama (comprimentos de onda: 589 nm para sódio e 767 nm para potássio), fazendo diluições adicionais, se necessárias.
¶ B.7.7.6 Cálculos e expressão dos resultados
Utilizando o método dos mínimos quadrados, interpolar na equação de regressão da curva de calibração de cada analito (concentrações dos padrões x sinais analíticos) o sinal analítico da amostra. Ajustar o resultado em função das diluições e massa de partida. Expressar o resultado de cada elemento com um número inteiro em “mg/kg” (se menor ou igual a 10000 mg/kg) ou em “g/kg”, com uma casa decimal (se maior que 10 g/kg).
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 969.23-1971, Sodium and potassium in seafood. Flame photometric method.
¶ B.8 Matéria mineral
¶ B.8.1 Princípio
O método se baseia na determinação gravimétrica do resíduo inorgânico, após remoção da água e queima da matéria orgânica, que é transformada em CO2, H2O e NO2, por aquecimento da amostra em forno-mufla à temperatura de 550 ºC ± 25 ºC. A matéria mineral (cinzas ou resíduo mineral fixo) obtida por calcinação corresponde ao resíduo inorgânico onde os elementos minerais se apresentam na forma de óxidos, sulfatos, fosfatos, silicatos e cloretos, dependendo das condições de incineração e da composição da amostra.
Alternativamente, o método ISO 5984 pode ser usado, dentro de seu escopo de aplicação.
¶ B.8.2 Campo de aplicação
Este método é aplicável aos produtos destinados à alimentação animal.
¶ B.8.3 Materiais e equipamentos
- Balança analítica, com resolução mínima de 0,001 g
- Cadinhos de porcelana
- Chapa aquecedora
- Dessecadores com sílica gel
- Espátulas
- Estufa de secagem
- Forno-mufla com tempo e temperatura programáveis
- Pinça metálica (tenaz) longa
- Pipetas
¶ B.8.4 Reagentes e soluções
- Peróxido de hidrogênio (H2O2, CAS 7722-84-1) 30%
¶ B.8.5 Procedimento de análise
- Aquecer os cadinhos em forno-mufla a 550 ºC por no mínimo 20 minutos.
- Quando a temperatura da mufla estiver entre 120 e 200 °C, retirar os cadinhos com a tenaz e acondicioná-los em dessecador até seu resfriamento.
- Pesar os cadinhos vazios, utilizando luvas de procedimento e/ou pinça.
- Adicionar cerca de 3 g de amostra a cada cadinho.
- Levar ao forno-mufla e fazer uma rampa de aquecimento, onde a temperatura seja aumentada gradativamente até 550 °C (tabela 1).
- Quando a temperatura da mufla estiver próxima a 200 °C, retirar os cadinhos com a tenaz e acondicioná-los em dessecador até seu resfriamento.
- Pesar os cadinhos com as cinzas em no máximo duas horas.
- Se houver pontos escuros (partículas de carbono) nas cinzas, umedecer as cinzas com gotas de água e adicionar gotas de peróxido de hidrogênio, com cuidado para não ocasionar perdas.
- Acondicionar os cadinhos em estufa a 103 °C até secagem do conteúdo e depois levá-los novamente à mufla por no mínimo duas horas a 550 ºC.

Nota 1: Antes da tara, os cadinhos não podem ser manuseados sem a utilização de luvas de procedimento, sendo ideal utilizar pinça (tenaz longa).
Nota 2: Os cadinhos com as cinzas não devem permanecer na mufla (< 200 °C) pois podem reabsorver umidade.
Nota 3: A mufla não deve ser aberta antes das amostras estarem completamente queimadas, pois poderão ocorrer chamas.
¶ B.8.6 Cálculo e expressão dos resultados
Calcular a matéria mineral em “g/kg” de acordo com a equação abaixo, reportando o valor da média das replicatas:
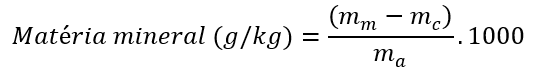
onde: mm = massa do cadinho + cinzas (g); mc = massa do cadinho (g); ma = massa de amostra (g).
¶ Bibliografia
- ISO 5984:2022 Animal feeding stuffs — Determination of crude ash.
¶ B.9 Nitrogênio total e proteína bruta
¶ B.9.1 Determinação por combustão (método Dumas)
¶ B.9.1.1 Princípio
O nitrogênio total é determinado pela combustão completa da amostra a alta temperatura (igual ou superior a 850 °C). Na presença de oxigênio puro (99,9%), os compostos nitrogenados gasosos são convertidos a óxidos de nitrogênio (NOx) que, em seguida, são reduzidos a nitrogênio elementar (N2), medido por um detector de condutividade térmica. A proteína bruta é calculada multiplicando-se a concentração de nitrogênio total por um fator de conversão apropriado. Alternativamente a este método, utilizar o procedimento descrito na norma ISO 16634-1.
¶ B.9.1.2 Campo de aplicação
Este método é aplicável aos produtos destinados à alimentação animal.
¶ B.9.1.3 Materiais e equipamentos
- Balança analítica com resolução mínima de 0,0001 g
- Cápsulas de estanho ou outro material adequado à pesagem da amostra, isento de nitrogênio
- Equipamento Dumas, equipado com forno capaz de manter temperatura igual ou superior a 850 °C, com detector de condutividade térmica e software para operação do instrumento, integração do sinal analítico e tratamento dos dados de análise
¶ B.9.1.4 Reagentes e soluções
- Ácido etilenodiamino tetra-acético (EDTA, C10H16N2O8, CAS 6381-92-6, material de referência certificado)
- Ácido aspártico (C4H7NO4, CAS 56-84-8), ácido L-glutâmico (C5H9NO4, CAS 56-86-0), ácido hipúrico (C9H9NO3, CAS 495-69-2), ácido nicotínico (C6H5NO2, CAS 59-67-6), cloridrato de L-lisina (C6H15ClN2O2, CAS 657-26-1), L-triptofano (C11H12N2O2, CAS 73-22-3) ou outras espécies químicas nitrogenadas apropriadas à verificação da exatidão do método
¶ B.9.1.5 Procedimento de análise
- Seguir as instruções do fabricante do equipamento para definição dos parâmetros instrumentais, otimização de métodos, calibração e operação.
- A cada batelada de amostras, analisar a quantidade de brancos necessária à estabilização do instrumento. A exatidão do método deve ser verificada com pelo menos um material de referência, distinto do utilizado na curva de calibração, analisado em triplicata a cada batelada de amostras. A média desta determinação deve variar no máximo 0,15 g de nitrogênio/100 g em relação ao valor teórico, com um desvio padrão de no máximo 0,15.
- Incluir, em cada batelada de amostras, a análise em triplicata de um material isento de nitrogênio (como celulose em pó ou sacarose).
- Fazer também três medições sucessivas de 100 mg de EDTA. Analisar como amostras desconhecidas. Consideram-se aceitáveis os resultados entre 9,5% e 9,6% de nitrogênio.
- Preparar curva de calibração com no mínimo cinco pontos e de forma que sua faixa linear abranja, no mínimo, de 0,2 g a 20 g de nitrogênio/100 g de amostra.
- Analisar as amostras em duplicata, pesando no mínimo 100 mg por réplica.
¶ B.9.1.6 Cálculos e expressão dos resultados
- Nitrogênio total: expressar o resultado em “g/kg”, com uma casa decimal.
- Proteína bruta: expressar o resultado em “g/kg” com uma casa decimal, multiplicando-se a concentração de nitrogênio total por 6,25 ou por outro fator de conversão apropriado, conforme o anexo D da norma ISO 16634-1:2008(E).
¶
¶ Bibliografia
- AOAC International. Official Methods of Analysis of AOAC International. AOAC Official Method 990.03-2002, Protein (crude) in animal feed. Combustion method.
- AOAC International. Official Methods of Analysis of AOAC International. AOAC Official Method 992.15-1992(1996), Crude protein in meat and meat products including pet foods. Combustion method.
- FAO. 2011. Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14.
- ISO 16634-1:2008(E). Food products — Determination of the total nitrogen content by combustion according to the Dumas principle and calculation of the crude protein content. Part 1: Oilseeds and animal feeding stuffs.
¶ B.9.2 Determinação por volumetria de neutralização (método Kjeldahl)
Utilizar o método descrito na norma ISO 1871. Expressar o resultado de nitrogênio total em “g/kg”, com uma casa decimal. Expressar o resultado de proteína bruta em “g/kg”, com uma casa decimal, multiplicando-se a concentração de nitrogênio total por 6,25 ou por outro fator de conversão apropriado.
¶ Bibliografia
- ISO 1871:2009 Food and feed products — General guidelines for the determination of nitrogen by the Kjeldahl method.
¶ B.10 Umidade e voláteis
¶ B.10.1 Rações, ingredientes para fabricação de rações e forrageiras parcialmente desidratadas
¶ B.10.1.1 Princípio
Este método determina a umidade dos alimentos para animais por gravimetria de volatilização. A matéria seca pode ser então calculada matematicamente. Se presentes nas amostras, compostos voláteis (como amônia, ácidos orgânicos e alguns ácidos graxos) também serão medidos. A amostra é submetida à secagem em condições especificadas e a perda de massa devida ao processo analítico é determinada por pesagem.
Nota: o ensaio deve ser executado imediatamente após a abertura das embalagens e preparo das amostras ou no máximo em 24 horas.
Alternativamente, o método AOAC 930.15-1930(1999) pode ser usado, dentro de seu escopo de aplicação.
¶ B.10.1.2 Campo de aplicação
Este método se aplica a amostras de rações, ingredientes para fabricação de rações e forrageiras parcialmente desidratadas, com matéria seca igual ou superior a 85% e baixos teores de ácidos orgânicos voláteis.
Amostras sólidas com umidade superior a 15 g/100 g (matéria seca inferior a 85%) deverão ser submetidas à pré-secagem descrita no item 7.11.4, antes da determinação de umidade.
Este método NÃO SE APLICA a amostras de rações com altos teores de açúcares, grãos inteiros, silagens, produtos lácteos para alimentação animal, óleos e gorduras animais e vegetais, sementes e frutos oleaginosos, que demandam métodos específicos (itens B.10.2 e B.10.3).
¶ B.10.1.3 Materiais e equipamentos
- Balança analítica com resolução mínima de 0,0001 g
- Cápsulas de metal inoxidável ou de vidro com tampas e dimensões aproximadas de 50 mm de diâmetro e 40 mm de profundidade, ou que permitam distribuição superficial da amostra na ordem de aproximadamente 0,3 g/cm2
- Dessecador com placa de metal ou de porcelana espessa perfurada e agente dessecante (cloreto de cálcio anidro ou sílica gel)
- Estufa de aquecimento elétrico, com regulação rápida da temperatura e convecção de ar
- Pinça tenaz
¶ B.10.1.4 Procedimento de análise
- Secar a cápsula com a tampa a 103 °C (± 2 °C) por 30 minutos (± 1 minuto). Com auxílio da pinça tenaz, retirar da estufa e acondicionar no dessecador, para resfriamento. Pesar.
- Pesar cerca de 5,00 g da amostra, distribuindo-a uniformemente sobre a cápsula.
- Acondicionar a cápsula destampada na estufa previamente aquecida a 103 °C (± 2 °C). Secar por quatro horas, contadas a partir do momento que a temperatura da estufa voltar a atingir 103 °C (± 2 °C). Abrir a estufa, tampar imediatamente a cápsula e acondicioná-la no dessecador, para resfriamento por 30 a 45 minutos. Pesar.
- Alimentos com teor de gordura superior a 50 g/100 g deverão ser submetidos a uma secagem suplementar a 103 °C (± 2 °C), por trinta minutos. A diferença entre as duas pesagens não deve exceder o valor correspondente a 0,1% de umidade.
- Quando necessária a pré-secagem, utilizar bandejas limpas e secas, de plástico ou de alumínio. Pesar o recipiente e registrar seu valor de massa. Transferir cerca de 300 g de amostra, distribuindo-a de forma uniforme. Acondicionar o recipiente contendo a amostra em estufa com ventilação forçada a uma temperatura estável inferior a 60 ºC, por um tempo de 48 a 72 horas, até que o material se apresente quebradiço, mas sem se escurecer, ou até peso constante (alternativamente, pode ser usado forno de micro-ondas doméstico, desde que o tempo e potência sejam ajustados às condições de pré-secagem). Retirar o recipiente da estufa e acondicionar em local protegido de poeira e umidade, deixando-o estabilizar por 15 minutos. Pesar e registrar o valor de massa. Após essa etapa, a amostra pré-seca deve ser moída e analisada.
- A diferença entre duas determinações paralelas, realizadas na mesma amostra, não deve exceder 0,2% do valor absoluto da umidade. Para alimentos úmidos para animais de estimação, essa diferença não pode exceder 0,5%.
¶ B.10.1.5 Cálculos e expressão dos resultados
- Sem pré-secagem:
O teor de umidade, expresso em “g/kg" com uma casa decimal, é calculado pela seguinte equação:
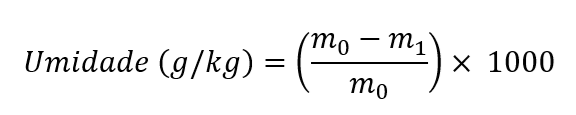
onde m0 = massa inicial, em gramas, da amostra; m1 = massa, em gramas, da amostra seca. O resultado deve ser expresso com duas casas decimais. Para expressar o resultado em g/kg, multiplicar por 10.
A matéria seca, expressa em “g/kg" com uma casa decimal, é calculada pela seguinte equação:
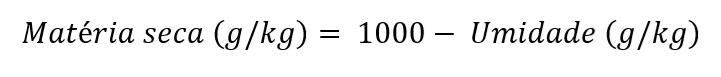
- Com pré-secagem:
A umidade de amostras submetidas à pré-secagem é expressa em “g/kg”, com uma casa decimal, sendo calculada pela equação:
onde m0 = massa inicial, em gramas, da amostra antes da pré-secagem; m1 = massa, em gramas, da amostra depois da pré-secagem.
¶ Bibliografia
- AOAC International. Official Methods of AOAC International. AOAC 930.15-1930(1999), Loss on drying (moisture) for feeds at 135 ºC for 2 hours).
- FAO. 2011. Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14. Rome.
- Regulamento de execução (UE) 2024/771 da Comissão de 29 de fevereiro de 2024 que altera o Regulamento (CE) n.° 152/2009 que estabelece os métodos de amostragem e análise para o controlo oficial dos alimentos para animais.
¶ B.10.2 Óleos e gorduras animais e vegetais, silagens, grãos inteiros, cereais e sêmolas
¶ B.10.2.1 Princípio
Este método determina a umidade dos alimentos para animais por gravimetria de volatilização. A matéria seca pode ser então calculada matematicamente. Se presentes nas amostras, compostos voláteis (como amônia, ácidos orgânicos e alguns ácidos graxos) também serão medidos. A amostra é submetida à secagem em condições especificadas e a perda de massa devida ao processo analítico é determinada por pesagem.
Nota: o ensaio deve ser executado imediatamente após a abertura das embalagens e preparo das amostras ou no máximo em 24 horas.
¶ B.10.2.2 Campo de aplicação
Este método se aplica a amostras de óleos e gorduras animais e vegetais, silagens, grãos inteiros, cereais e sêmolas, preparadas para análise conforme item 7.1 deste manual.
¶ B.10.2.3 Materiais e equipamentos
- Balança analítica com resolução mínima de 0,0001 g
- Cápsulas de metal inoxidável ou de vidro com tampas e dimensões aproximadas de 50 mm de diâmetro e 40 mm de profundidade, ou que permitam distribuição superficial da amostra na ordem de aproximadamente 0,3 g/cm2
- Dessecador com placa de metal ou de porcelana espessa perfurada e agente dessecante (cloreto de cálcio anidro ou sílica gel)
- Estufa de aquecimento elétrico, com regulação rápida da temperatura e convecção de ar
- Pinça tenaz
¶ B.10.2.4 Procedimento de análise
- Secar a cápsula com a tampa na estufa regulada à temperatura de secagem correspondente (tabela 1), por 30 minutos (± 1 minuto). Com auxílio da pinça tenaz, retirar da estufa e acondicionar no dessecador, para resfriamento. Pesar.
- Pesar uma alíquota da amostra conforme tabela 1, distribuindo-a uniformemente sobre a cápsula.
- Acondicionar a cápsula destampada na estufa previamente aquecida à temperatura e pelo tempo especificados na tabela 1, contado a partir do momento que a temperatura da estufa volte a atingir a especificação. Abrir a estufa, tampar imediatamente a cápsula e acondicioná-la no dessecador, para resfriamento por 30 a 45 minutos. Pesar.
- Repetir a operação acima, com períodos de secagem de 30 minutos, até que o peso da amostra seca seja constante.
- A diferença entre duas determinações paralelas, realizadas na mesma amostra, não deve exceder 0,2% do valor absoluto da umidade.

¶ B.10.2.5 Cálculos e expressão dos resultados
O teor de umidade, expresso em “g/kg", com uma casa decimal, é calculado pela seguinte equação:
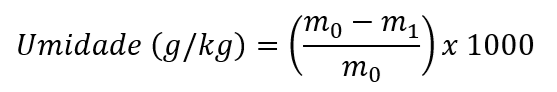
onde m0 = massa inicial, em gramas, da amostra; m1 = massa, em gramas, da amostra seca.
A matéria seca, expressa em “g/kg", com uma casa decimal, é calculada pela seguinte equação:
¶ Bibliografia
- FAO. 2011. Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14. Rome.
- Regulamento de execução (UE) 2024/771 da Comissão de 29 de fevereiro de 2024 que altera o Regulamento (CE) n.° 152/2009 que estabelece os métodos de amostragem e análise para o controlo oficial dos alimentos para animais.
¶ B.10.3 Açúcares, alfarroba, bagaço, melaço, produtos cereais hidrolisados, germes de malte, rações com teores de sacarose ou lactose superiores a 4% e produtos lácteos para alimentação animal
¶ B.10.3.1 Princípio
Este método determina a umidade dos alimentos para animais por gravimetria de volatilização. A matéria seca pode ser então calculada matematicamente. Se presentes nas amostras, compostos voláteis (como amônia, ácidos orgânicos e alguns ácidos graxos) também serão medidos. A amostra é submetida à secagem em condições especificadas e a perda de massa devida ao processo analítico é determinada por pesagem.
Nota: o ensaio deve ser executado imediatamente após a abertura das embalagens e preparo das amostras ou no máximo em 24 horas.
¶ B.10.3.2 Campo de aplicação
Este método se aplica a amostras de açúcares, alfarroba, bagaço, melaço, produtos cereais hidrolisados, germes de malte, rações com teores de sacarose ou lactose superiores a 4% e produtos lácteos para alimentação animal.
¶ B.10.3.3 Materiais e equipamentos
- Balança analítica com resolução mínima de 0,0001 g
- Cápsulas de metal inoxidável ou de vidro com tampas e dimensões aproximadas de 50 mm de diâmetro e 40 mm de profundidade, ou que permitam distribuição superficial da amostra na ordem de aproximadamente 0,3 g/cm2
- Dessecador com placa de metal ou de porcelana espessa perfurada e agente dessecante (cloreto de cálcio anidro ou sílica gel)
- Estufa de vácuo com aquecimento elétrico regulável, munida de bomba de óleo e dispositivo de introdução de ar quente seco
- Pinça tenaz
¶ B.10.3.4 Procedimento de análise
- Secar a cápsula com a tampa a uma temperatura entre 80 °C e 85 ºC por 30 minutos (± 1 minuto). Com auxílio da pinça tenaz, retirar da estufa e acondicionar no dessecador, para resfriamento. Pesar.
- Pesar cerca de 5,00 g da amostra, distribuindo-a uniformemente sobre a cápsula.
- Acondicionar rapidamente a cápsula destampada na estufa de vácuo previamente aquecida a uma temperatura entre 80 °C e 85 ºC. Regular a pressão a 100 Torr e deixar a amostra secar por quatro horas, sob corrente de ar quente e seco, contadas a partir do momento que a temperatura da estufa voltar a atingir a temperatura de 80 °C a 85 ºC. Reduzir a pressão no interior da estufa até a pressão atmosférica. Abrir a estufa, tampar imediatamente a cápsula e acondicioná-la no dessecador, para resfriamento por 30 a 45 minutos. Pesar.
- Efetuar secagem complementar de 30 minutos na estufa de vácuo a uma temperatura entre 80 °C e 85 ºC e pesar novamente, de forma que a diferença entre as duas pesagens não exceda 0,1% do valor de umidade.
- A diferença entre duas determinações paralelas, realizadas na mesma amostra, não deve exceder 0,2% do valor absoluto da umidade. Para alimentos úmidos para animais de estimação, essa diferença não pode exceder 0,5%.
¶ B.10.3.5 Cálculos e expressão dos resultados
1. O teor de umidade, em “g/kg”, é calculado pela seguinte equação:
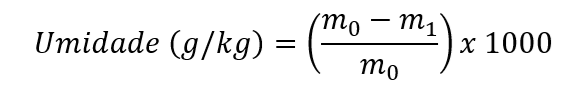
onde m0 = massa inicial, em gramas, da amostra; m1 = massa, em gramas, da amostra seca. O resultado deve ser expresso com uma casa decimal.
2. A matéria seca é calculada pela seguinte equação:
¶ Bibliografia
- FAO. 2011. Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14. Rome.
- Regulamento de execução (UE) 2024/771 da Comissão de 29 de fevereiro de 2024 que altera o Regulamento (CE) n.° 152/2009 que estabelece os métodos de amostragem e análise para o controlo oficial dos alimentos para animais.
¶ B.11 Umidade e voláteis (por secagem por micro-ondas e/ou infravermelho) e gordura total (por ressonância magnética nuclear)
¶ B.11.1 Princípio
Este método baseia-se na determinação gravimétrica da umidade após evaporação da água contida na amostra, promovida pela energia de micro-ondas e/ou infravermelho, utilizados como fonte de radiação, seguida da determinação direta da gordura total por ressonância magnética nuclear em domínio de tempo de baixa resolução.
A umidade é expressa como fração mássica, sendo determinada automaticamente pela balança eletrônica do instrumento por diferença de massa (antes e depois da secagem da amostra). Se presentes, compostos voláteis também serão medidos.
A gordura total é determinada por energia de radiofrequência pulsada, submetendo-se a amostra seca a um campo magnético estático de 0,55 T. O sinal analítico resultante da atividade total de prótons da gordura é convertido em fração mássica.
¶ B.11.2 Campo de aplicação
Este método se aplica à determinação quantitativa da umidade e voláteis e da gordura total em amostras de produtos destinados à alimentação animal.
¶ B.11.3 Materiais e equipamentos
- Estação de trabalho com suporte para rolo de filme “Trac” e prensa manual para tubos de ressonância magnética nuclear
- Filme “Trac” picotado para embalagem da amostra para determinação de gordura no módulo Oracle (CEM Corporation, part no. 159875). Quando um novo rolo de filme for aberto, fazer a leitura do código de barras antes de abrir a embalagem.
- Placas de fibra de fibra de vidro para suporte da amostra para determinação de umidade no módulo Smart 6 (CEM Corporation, part no. 200150). Depois de aberta, a embalagem das placas de fibra de vidro deve ser preferencialmente mantida em dessecador, para evitar absorção de umidade.
- Prensa manual para placas de fibra de vidro
- Sistema composto por módulo analisador de umidade (Smart 6, CEM Corporation) e por espectrômetro de ressonância magnética nuclear (módulo Oracle, CEM Corporation) para análise de gordura total, podendo ser utilizados de forma isolada ou combinada
- Tubos de polietileno para análise no RMN
¶ B.11.4 Procedimento de análise
¶ B.11.4.1 Determinação da umidade e voláteis
- Ligar o analisador de umidade e selecionar o método instrumental apropriado para a matriz a ser analisada.
- Abrir a tampa do instrumento e colocar uma (se necessário, duas) placa(s) de fibra de vidro sobre a balança.
- Fechar a tampa e tarar.
- Digitar o número de identificação e transferir de 1,00 g a 2,00 g da amostra para o centro da parte rugosa de uma placa de suporte. Espalhar. O valor da massa de amostra será gravado pelo instrumento. Se necessário, cobrir com outra placa tarada, de forma que seu lado rugoso entre em contato com a amostra. Se utilizadas duas placas, prensar suavemente as placas para que a amostra se espalhe em camada fina e uniforme e colocá-las novamente sobre a balança.
- Fechar a tampa do instrumento e iniciar a análise.
- A umidade será calculada automaticamente quando a pesagem atingir valor constante.
¶ B.11.4.2 Determinação da gordura total
- Inicializar o espectrômetro.
- Remover do analisador de umidade as placas de fibra de vidro contendo a amostra seca e envolvê-las com uma folha de filme “Trac”, respeitando o tempo máximo de 45 segundos entre o fim da análise de umidade e sua inserção no espectrômetro, para evitar a absorção de água.
- Inserir a embalagem no tubo de polietileno, usando a prensa para compacta-la no fundo (parte fosca do tubo).
- Pressionar “Start” para fechamento da válvula solenoide do espectrômetro e inserir o tubo contendo a amostra na câmara de análise.
- Após o tempo de condicionamento da amostra à temperatura de análise, o resultado será exibido automaticamente no instrumento.
Nota: A temperatura da sala analítica deve estar entre 15 °C e 30 °C, sem sofrer variação superior a 5 °C.
¶ B.11.5 Cálculo e expressão dos resultados
Os resultados de umidade e de gordura total deverão ser expressos em “g/kg”, com uma casa decimal.
¶ Bibliografia
- AOAC International. Official Methods of Analysis of AOAC International. AOAC Official Method AOAC 2008.06-2008, Moisture and fat in meats. Microwave and nuclear magnetic resonance analysis.
¶ B.12 Umidade, fibra bruta, gordura total e proteína bruta (composição bromatológica) por espectroscopia no infravermelho próximo
¶ B.12.1 Princípio
A composição química da amostra é medida por espectroscopia no infravermelho próximo (NIRS), técnica vibracional que corresponde à faixa de comprimento de onda do espectro eletromagnético de 750 nm a 2500 nm (números de onda de 13330 cm-1 a 4000 cm-1). A interação da radiação nessa faixa de comprimento de onda (ou em segmentos selecionados dessa região) com as espécies químicas é medida por meio da relação da refletância (ou da transmitância) difusa da amostra e a intensidade da radiação para um material de referência (spectralon ou cerâmica). Os dados espectrais das amostras são coletados e transformados em concentrações de umidade, gordura total, proteína bruta e fibra bruta através de modelos de calibração desenvolvidos com amostras representativas, de acordo com as normas ASTM 1655-17 e ISO 12099:2017. A composição mineral é calculada por diferença. Por se tratar de uma técnica vibracional, a maioria das vantagens da espectroscopia NIRS advém da possibilidade de utilização de amostras intactas inseridas diretamente no equipamento, sem a necessidade de pré-tratamento.
¶ B.12.2 Campo de aplicação
Este método é aplicável a amostras de rações e ingredientes para fabricação de rações para predição de sua composição bromatológica em termos de umidade, proteína bruta, fibra bruta, matéria mineral e gordura total.
¶ B.12.3 Materiais e equipamentos
¶ B.12.3.1 Espectrômetros
Para a maioria das aplicações envolvendo matrizes complexas, instrumentos NIRS de varredura, dispersivos ou baseados em interferômetro oferecem melhor desempenho, devido à estabilidade e reprodutibilidade da instrumentação e à maior quantidade de dados espectrais fornecidos para a construção de modelos de calibração multivariados. Esses dados permitem maior flexibilidade de calibração e opções adicionais para a seleção de áreas espectrais menos sensíveis a deslocamentos de bandas e ruídos dentro do sinal espectral. Além disso, permitem maior precisão de comprimento de onda/frequência entre instrumentos, devido às técnicas internas de padronização de comprimento de onda/frequência, com a possibilidade de correções espectrais geradas por computador.
Os espectrômetros NIRS devem ser equipados com um sistema de diagnóstico para teste de ruído fotométrico e reprodutibilidade e precisão do comprimento de onda/número de onda (para espectrofotômetros de varredura). O instrumento deve medir um volume ou superfície de amostra suficientemente grande para eliminar qualquer influência significativa de não homogeneidade derivada da composição química ou das propriedades físicas da amostra. O comprimento do caminho da amostra (espessura da amostra) nas medições de transmitância deve ser otimizado de acordo com as recomendações do fabricante em relação à intensidade do sinal para obter linearidade e máxima relação sinal/ruído. Nas medições de refletância, uma janela de quartzo ou outro material apropriado para eliminar os efeitos de secagem deve, preferencialmente, cobrir a camada superficial da amostra em interação.
¶ B.12.3.2 Requisitos quimiométricos
A quimiometria é fundamental para o desenvolvimento de modelos qualitativos e quantitativos de calibração multivariada por meio de algoritmos matemáticos e estatísticos. O avanço dos softwares quimiométricos possibilitou o implemento de técnicas de análises estatísticas multivariadas complexas de fácil aplicação, destacando-se a análise de componentes principais (PCA) e a regressão de quadrados mínimos parciais (PLS), amplamente utilizadas em análises qualitativas e quantitativas, respectivamente.
As normas ASTM 1655-17 e ISO/FDIS 12099:2017(E) fornecem informações para a correta a seleção de amostras representativas, calibração multivariada, validação e manutenção dos modelos quimiométricos para uso de NIRS.
¶ B.12.4 Procedimento de análise
¶ B.12.4.1 Construção de novos modelos de calibração
¶ B.12.4.1.1 Amostragem
Todas as amostras devem ser mantidas em condições que não alterem sua composição desde o momento da coleta até o início do procedimento no laboratório.
O laboratório deve receber as amostras representativas, com a maior variabilidade amostral possível, considerando para um mesmo tipo de material as variações em relação às espécies/cultivares, sazonalidade e local de coleta, para que apresentem uma faixa ampla de concentrações dos analitos de interesse. As amostras não podem ter sido danificadas ou alteradas durante o transporte ou armazenamento.
A seleção das amostras deve:
- Incluir amostras que englobem todos as propriedades químicas que se esperam estar presentes nas amostras que serão analisadas pelo modelo, assegurando que essas análises envolvem apenas interpolação do modelo, ou seja, que não ocorra previsões com extrapolação.
- Conter amostras em que a faixa de variação na concentração dos componentes químicos exceda a faixa de variação esperada para as amostras que devem ser analisadas usando o modelo, garantindo assim que as análises envolvam apenas interpolação do modelo.
- Conter amostras em que as concentrações dos componentes químicos estejam uniformemente distribuídas (distribuição retangular) sobre sua faixa total de variação (faixa de trabalho de calibração).
- Conter um número suficiente de amostras para definir estatisticamente a relação entre as variáveis espectrais e a concentração ou propriedade do componente a ser modelada.
- Para a validação de um modelo multivariado o conjunto de amostras de validação deve obedecer às especificações 1, 2, 3 e 4 descritas para o conjunto de calibração.
- Conter um número suficiente de amostras para testar estatisticamente a relação entre as variáveis espectrais e as concentrações ou propriedades dos analitos que foram modeladas.
Apenas as amostras cujas análises são interpolações do modelo devem ser usadas no procedimento de validação. Dessa forma, a faixa de concentração dos analitos nas amostras de calibração, isto é, a amplitude e o desvio padrão desses valores em relação às amostras de validação deve ser pelo menos 95% das amostras de calibração e os valores de concentração dos analitos nas amostras de validação. Além disso, os valores de concentração devem estar distribuídos o mais uniformemente quanto possível ao longo da faixa trabalho de calibração; caso contrário, amostras adicionais devem ser incluídas na validação do modelo.
¶ B.12.4.1.2 Coleta dos dados espectrais das amostras de calibração e validação
- A coleta dos espectros das amostras de calibração/validação deve seguir as instruções do fabricante do instrumento.
- As amostras preparadas para o ensaio devem atingir temperatura dentro da faixa incluída no procedimento de validação.
- A matriz de dados espectrais deve ser arquivada de forma organizada e rastreável em software quimiométrico para posterior associação aos resultados obtidos pelos métodos de referência e construção/validação dos modelos multivariados.
¶ B.12.4.1.3 Resultados de referência para o conjunto de amostras de calibração, validação e validação externa
- Os métodos de referência devem ser validados e atender aos requisitos de garantia da validade dos resultados e ao escopo de matrizes e analitos previstos nos modelos de calibração para análise por NIRS.
- As figuras de mérito da validação como exatidão e precisão (repetibilidade, precisão intermediária e reprodutibilidade) devem ser observadas na escolha do método de referência a ser utilizado para a execução das análises do conjunto de amostras a ser utilizado na calibração do modelo NIRS.
Nota: se os resultados analíticos forem obtidos de métodos de referência que possuem baixa precisão e exatidão, poderão impactar diretamente na incerteza, na robustez dos modelos de calibração NIRS e nos dados por eles gerados.
¶ B.12.4.1.4 Validação em espectroscopia NIRS
Parâmetros de mérito para modelos de calibração multivariada desenvolvidos a partir de mínimos quadrados parciais (PLS – partial least squares) e espectroscopia NIRS: indicadores de desempenho de que o modelo desenvolvido apresenta habilidade preditiva satisfatória para uso em rotina analítica:
- Precisão: a precisão está relacionada ao erro aleatório e expressa o grau de concordância para os resultados de repetidas avaliações em uma mesma amostra e sob as mesmas condições analíticas. Assim como a calibração univariada, numa calibração por NIR/PLS a precisão pode ser estimada em três níveis: repetibilidade, precisão intermediária e reprodutibilidade. Os parâmetros de mérito de validação em NIRS que estão associados à precisão são o SECV (erro padrão de validação cruzada), SEC (erro padrão de calibração) e SEP (erro padrão de predição). O SEP deve ser relacionado ao SEC (ou SECV) para verificar a validade e consistência do modelo de calibração para o conjunto de validação selecionado.
- Exatidão: grau de concordância entre um resultado de ensaio e o valor de referência aceito. Quando aplicado a um conjunto de resultados de ensaio, a exatidão inclui a combinação de componentes aleatórios e sistemáticos comuns ou componente de tendência. A exatidão em calibração PLS/NIR pode ser avaliada através de um conjunto de parâmetros – incluindo o ajuste e os erros – representados pelo erro médio quadrático de calibração (RMSEC), erro médio quadrático de validação externa (RMSEP) e erro médio quadrático de validação cruzada (RMSECV). Esses erros são bons indicadores da adequada dimensionalidade do modelo, ou seja, da escolha adequada do número de variáveis latentes (VLs), associando a estabilidade desses parâmetros para definir o número de VLs. Nesse sentido, modelos com dimensionalidade adequada apresentam erros similares, com resultados próximos (na situação ideal, erros iguais) para os parâmetros RMSEC e RMSEP. É comum que o RMSECV seja ligeiramente superior ao RMSEC que, por sua vez, pode ser também ligeiramente superior ao RMSEP. Os resultados de RMSEC, RMSEP e RMSECV são indicadores da tendência ou do viés (bias, erro sistemático), sendo geralmente considerados em conjunto com o ajuste na avaliação da exatidão de modelos NIR/PLS.
- Bias: erro sistemático calculado pela diferença entre a média da população e o valor verdadeiro e avaliado a partir de teste ‘t’ nas amostras de predição (validação externa).
- Relação de desempenho do desvio (RPD – residual prediction deviation): útil para comparar modelos em diferentes conjuntos de dados, ou em termos absolutos. RPD é a razão entre a variação natural das amostras e o tamanho dos erros prováveis que ocorrem durante a predição. A RPD é estimada para os conjuntos de calibração (RPDcal) e validação externa (RPDval), de acordo com as equações abaixo:
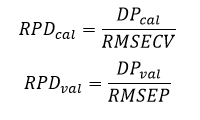
onde DPcal corresponde ao desvio padrão dos valores de referência do conjunto de calibração; DPval corresponde ao desvio padrão dos valores de referência do conjunto de validação externa.
De acordo com esse parâmetro, modelos com boa capacidade preditiva devem ter um valor de RPD acima de 2,4, enquanto valores entre 2,4 e 1,5 são considerados satisfatórios. Modelos com RPD menor que 1,5 não devem ser usados.
- Linearidade: considerando que as variáveis espectrais NIR são decompostas pelas variáveis latentes no PLS, a linearidade dos modelos NIR/PLS é avaliada de forma qualitativa a partir do gráfico dos resíduos. Uma distribuição aleatória e homocedástica para os resíduos é um indicativo de que os dados se ajustam ao modelo linear.
¶ B.12.4.2 Análise de amostras de rotina
¶ B.12.4.2.1 Preparo de amostra
O preparo das amostras deve ser feito da mesma forma que o preparo das amostras utilizadas na validação/calibração do método para cada matriz específica.
¶ B.12.4.2.2 Medição
Seguir as instruções do fabricante do instrumento e das calibrações associadas. As amostras preparadas para o ensaio devem atingir temperatura dentro da faixa incluída no procedimento de validação.
¶ B.12.4.2.3 Avaliação do resultado
Para que os resultados sejam válidos, devem estar dentro do intervalo do modelo de calibração. Os resultados obtidos por NIRS para as amostras de rotina cujos valores sejam considerados discrepantes espectrais (outliers) não serão aceitos e deverão ser submetidos à análise por outras técnicas, de acordo com os métodos deste manual.
¶ B.12.4.2.4 Avaliação da estabilidade do instrumento
¶ B.12.4.2.4.1 Amostras de controle
Pelo menos uma amostra de controle deve ser medida durante a rotina analítica para verificar estabilidade do hardware do instrumento e para detectar qualquer mau funcionamento. O material deve ser estável e, tanto quanto possível, de composição química semelhante às amostras que de rotina. O(s) parâmetro(s) medido(s) devem ser estáveis e, tanto quanto possível, idênticos ou quimicamente próximos do analito nas amostras. A variação diária registrada deve ser plotada em cartas de controle e checada quanto a perfis ou tendências significativas.
¶ B.12.4.2.4.2 Diagnóstico do instrumento
A exatidão e a precisão do comprimento de onda/número de onda devem ser verificadas pelo menos uma vez por semana, ou com mais frequência se recomendado pelo fabricante do instrumento. A verificação do ruído do instrumento também deve ser realizada semanalmente, ou nos intervalos recomendados pelo fabricante.
¶ B.12.5 Cálculos e expressão dos resultados
Expressar os resultados obtidos para umidade, fibra bruta, gordura total, matéria mineral e proteína bruta em “g/kg” e uma casa decimal.
Os resultados obtidos por NIRS para as amostras de rotina cujos valores sejam considerados discrepantes espectrais (outliers) não serão aceitos e deverão ser submetidos à análise por outros métodos constantes deste manual.
Os resultados obtidos por NIRS que estejam em não conformidade com os níveis de garantia declarados para as amostras de rotina deverão ser confirmados pelos métodos constantes deste manual para aquele(s) parâmetro(s).
¶ B.12.6 Glossário
¶ B.12.6.1 Técnicas de calibração
- Alavancagem (leverage): medida de quão longe uma amostra está do centro do espaço populacional definido por um modelo. Amostras com alta alavancagem têm grande influência no modelo. A idade da alavancagem é calculada medindo a distância entre um ponto projetado e o centro do modelo.
- Análise de componentes principais (PCA): é uma forma de compactação de dados. Para um conjunto de amostras, ela trabalha apenas com os dados X (dados espectrais) e encontra componentes principais (fatores) de forma que cada PC expresse a variação máxima dos dados a qualquer momento, sem estar correlacionada com nenhum outro PC. O primeiro PC expressa tanto quanto possível a variabilidade dos dados originais. Seu efeito é então subtraído dos dados X de um novo PC derivado novamente, expressando a máxima variabilidade possível dos dados restantes. Podem ser derivados tantos PCs quantos forem os pontos de dados no espectro ou amostras do conjunto de dados, mas os principais efeitos nos espectros estão concentrados em apenas alguns dos primeiros PCs e, portanto, a quantidade de dados que precisa ser considerada é drasticamente reduzida. O PCA produz dois novos conjuntos de variáveis a cada estágio: As pontuações do PC representam a resposta de cada amostra em cada PC; carregamentos de PC representam a importância de cada ponto de dados nos espectros originais em relação ao PC. PCA pode ter várias aplicações, como a interpretação espectral, embora seja mais utilizada para a identificação de valores espectrais discrepantes.
- Conjunto de teste: quando um modelo de regressão é testado, é qualquer conjunto de amostras que exclua aquelas usadas para desenvolver a calibração.
- Conjunto de teste independente: consiste em amostras provenientes de uma região geográfica diferente, de novo estabelecimento industrial ou que foram coletadas posteriormente às amostras usadas para criar e validar um modelo de regressão. Essas amostras formam um teste “verdadeiro” de um modelo de predição.
- Conjunto de validação: amostras usadas para validar ou comprovar uma calibração, que têm as mesmas características daquelas selecionadas para calibração. Frequentemente alternadas ou amostras “enésimas” (classificadas em ordem do constituinte de interesse) são alocadas para a calibração e conjuntos de dados de validação do mesmo conjunto de amostras.
- Distância de Mahalanobis: é a distância no espaço PC entre um ponto de dados e o centro do espaço do PC (veja “valor h”, abaixo). É uma medição não linear. No espaço PC, os conjuntos de amostras geralmente formam uma distribuição em forma de curva. O elipsoide que melhor representa a distribuição de probabilidade do conjunto pode ser estimada pela matriz de covariância das amostras. A distância de Mahalanobis é simplesmente a distância do ponto de teste ao centro de massa dividido pela largura do elipsoide na direção do ponto de teste.
- Modelo multivariado: qualquer modelo onde vários valores de X são usados para predizer uma ou mais variáveis Y.
- Outliers (valores discrepantes): são pontos em qualquer conjunto de dados que por demonstração estatística apresentam valores que estão fora da distribuição esperada. Para dados NIRS, tanto os valores X (dados espectrais) quanto os valores Y (dados de referência) podem apresentar valores discrepantes. Os outliers X podem estar relacionados a falhas instrumentais ou a um tipo de amostra muito diferente das outras amostras, não incluído no conjunto de calibração original. Outliers Y estão relacionados a erros nos dados de referência, como um erro de transcrição de dados ou na obtenção do valor pelo método de referência.
- Pontuações/gráficos de pontuação: gráficos onde as pontuações em um fator PC ou PLS são plotadas em relação às pontuações de outro fator PC ou PLS. Mais útil se a identidade da amostra ou os valores de concentração forem usados para identificar cada ponto do gráfico. Podem então ser vistos perfis que não são óbvios de se identificar a partir dos dados brutos.
- Regressão do componente principal (PCR): usa as pontuações de cada PC como regressores em uma regressão linear múltipla contra valores Y que representam a composição das amostras. Como cada PC é ortogonal a todos os outros PCs, as pontuações formam um conjunto de dados não correlacionados com propriedades melhores do que os espectros originais. Embora seja possível selecionar uma combinação de PCs para regressão com base em quão bem cada PC se correlaciona com o constituinte de interesse, a maioria dos softwares comerciais força a regressão a usar todos os PCs até o PC mais alto selecionado para o modelo (abordagem “de cima para baixo”). Quando usados no NIRS, os coeficientes de regressão no espaço do PC são convertidos de volta para um modelo de predição, usando todos os pontos de dados em um intervalo de comprimento de onda.
- Regressão linear múltipla (MLR): usa uma combinação de várias variáveis X para predizer uma única variável Y. No NIRS, os valores X são valores de absorbância em comprimentos de onda selecionados no infravermelho próximo ou variáveis derivadas, como pontuações PCA ou PLS.
- Redes neurais artificiais (RNA): técnica de modelagem não linear baseada em sistemas neurais biológicos. A rede é inicialmente “treinada” pelo fornecimento de um conjunto de dados com vários valores X (variáveis espectrais ou derivadas, como pontuações PCA) e valores de referência Y. Durante o processo de treinamento, a arquitetura da rede pode ser modificada e aos neurônios podem ser atribuídos coeficientes de ponderação para entradas e saídas de forma a produzir as melhores predições possíveis dos valores dos parâmetros. Redes Neurais exigem muitos dados em treinamento.
- Regressão parcial de mínimos quadrados (PLS): é uma técnica analítica que, como PCA, é uma forma de compactação de dados. Com o PLS, a regra usada para derivar os fatores é que cada fator maximiza a covariância entre os dados Y e todas as combinações lineares possíveis dos dados X. PLS é, portanto, um equilíbrio entre variância e correlação com cada fator sendo influenciado por ambos os efeitos. Os fatores PLS estão, portanto, mais diretamente relacionados com a variabilidade dos valores Y do que os PCs. PLS produz três novas variáveis, carregando pesos (que não são ortogonais entre si), cargas e pontuações que são ambas ortogonais. Os modelos PLS são produzidos por regressão das pontuações PLS em relação aos valores Y. Tal como acontece com PCR, quando usado em NIRS, os coeficientes de regressão no espaço PLS são geralmente convertidos de volta para um modelo de predição usando todos os pontos de dados no espaço de comprimento de onda.
- Residual: é a diferença entre um valor de referência e o valor predito por um modelo de regressão, usada no cálculo de estatísticas de regressão.
- Sobreajuste: a adição de dois termos de regressão em uma regressão linear múltipla. Como resultado, quando amostras que não estão no conjunto de calibração são preditas, estatísticas como RMSEP ou SEP são muito mais pobres do que o esperado.
- Validação cruzada: é um método de geração de estatísticas de predição onde, repetidamente, um subconjunto de amostras é removido da população de calibração e o modelo é calculado com as amostras restantes e os resíduos calculados com o subconjunto de validação. Quando esse processo tiver sido executado várias vezes, as estatísticas de predição são calculadas sobre todos os resíduos.
- Validação cruzada completa: omite uma amostra por vez e é executada n vezes (onde há n amostras de calibração). Quando um subconjunto maior é removido, o ciclo de validação cruzada é executado pelo menos oito vezes antes do cálculo estatístico. Finalmente, o modelo é calculado usando todas as amostras de calibração. A validação cruzada deve ser usada com cautela. Em primeiro lugar, estatísticas de validação cruzada tendem a ser otimistas quando comparadas com aquelas de um conjunto de teste. Em segundo lugar, deve-se tomar cuidado se houver alguma duplicação nos dados de calibração (por exemplo, uma mesma amostra analisada em vários instrumentos ou em momentos diferentes) para sempre atribuir todas as cópias da mesma amostra ao mesmo segmento de validação cruzada, caso contrário, estatísticas muito otimistas são produzidas.
- Valor h: em alguns softwares, a distância de Mahalanobis é referida como “valor h global” e a detecção de valores discrepantes dependem de quantos desvios padrão do valor h a amostra está em relação ao centro. Uma segunda medida (“vizinhança h”) é a distância no espaço PC entre um ponto de dados e seus “n” vizinhos mais próximos e indica o quanto uma amostra está isolada ou em uma parte bem povoada da distribuição.
¶ B.12.6.2 Termos estatísticos empregados em espectroscopia NIRS
- RMSEP (raiz do erro quadrático médio de predição): é uma expressão da diferença média entre os valores de referência e aqueles preditos por um modelo de regressão quando aplicada a um conjunto de amostras não incluídas na derivação do modelo. RMSEP inclui qualquer viés das predições.
- RMSECV (erro quadrático médio de validação cruzada): é uma expressão da diferença média entre os valores preditos e os valores de referência para o subconjunto de amostras selecionadas como amostras de predição durante o processo de validação cruzada (consulte “validação cruzada” acima). RMSECV inclui qualquer viés das predições.
- SEC (erro padrão de calibração): para qualquer modelo de calibração, o SEC é uma expressão da diferença média entre os valores preditos e os valores de referência para amostras usadas para derivar o modelo. Nesta e nas estatísticas subsequentes, a expressão da diferença média refere-se à raiz quadrada da soma dos quadrados dos valores residuais dividida pelo número de valores corrigidos para graus de liberdade, ou seja, o limite que cobre 68% dos erros. Isso é necessário porque alguns resíduos são positivos, outros negativos.
- SECV (erro padrão de validação cruzada): para um modelo de calibração, o SECV é uma expressão da diferença média corrigida pelo viés entre os valores preditos e os valores de referência para o subconjunto de amostras de predição selecionadas durante o processo de validação cruzada (veja ‘’validação cruzada” acima).
- SEP (erro padrão de predição): SEP é uma expressão da diferença média corrigida pelo viés entre os valores preditos e os valores de referência preditos por um modelo de regressão quando aplicada a um conjunto de amostras não incluídas na derivação do modelo.
- Viés (bias): é a diferença entre o valor médio de referência e o valor médio predito pelo modelo NIR.
¶ Bibliografia
- ASTM - American Society for Testing and Materials. Standard practices for infrared multivariate quantitative analysis, E1655-17, ASTM International, West Conshohocken, PA, 2017.
- Compêndio Brasileiro de Alimentação Animal. 2023. Uso da espectroscopia no infravermelho próximo como método de análise química. São Paulo: Sindicato Nacional da Indústria da Alimentação Animal (2023), cap. 12.
- FAO. 2011. Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14. Rome.
- ISO 12099:2017 Animal feeding stuffs, cereals and milled cereal products — Guidelines for the application of near infrared spectrometry.
¶ C. Métodos microscópicos
¶ C.1 Detecção de subprodutos de origem animal
¶ C.1.1 Princípio
Este método baseia-se na detecção e identificação microscópica de subprodutos de origem animal conforme tamanho, forma, cor, textura, dureza, brilho e outras características de seus fragmentos, com auxílio de testes físico-químicos qualitativos.
¶ C.1.2 Campo de aplicação
Este método visa à detecção de farinha de carne e ossos, farinha de penas hidrolisadas, farinha de vísceras de aves, farinha de peixe e farinha de sangue (ou de seus componentes) em misturas de ingredientes para alimentação de ruminantes, podendo também ser aplicado a alimentos destinados a outros animais, para a identificação de materiais estranhos ao seu processo de fabricação.
¶ C.1.3 Materiais e equipamentos
- Alicates
- Almofariz e pistilo
- Balança analítica
- Bandejas
- Béqueres
- Copos de sedimentação (cálice ou cápsulas) de vidro borossilicato ou porcelana e capacidade de 250 mL
- Colheres de cabo longo ou espátulas
- Equipamento de sucção
- Estiletes
- Frascos de coloração âmbar com conta-gotas
- Frasco kitasato com capacidade de 2 L
- Funil de Büchner com diâmetro mínimo de 12 cm
- Funis de separação (250 mL ou 500 mL), com diâmetro do orifício de drenagem e torneira aumentados
- Lâminas e lamínulas
- Microscópio estereoscópio, com aumento mínimo de 40 vezes
- Microscópio de fluorescência
- Papel de filtro
- Peneira de plástico, com aberturas de 0,1 mm a 0,2 mm
- Placas de Petri
- Pinças pontiagudas
- Provetas com capacidade de 50 mL e 100 mL
- Tamises de malhas com aberturas de no mínimo 1,4 mm, 1,0 mm e 0,5 mm
- Tubos de ensaio
¶ C.1.4 Reagentes e soluções
- Acetato de chumbo 2% (Pb(C2H3O2)2, CAS 301-04-2) em hidróxido de sódio 10% (NaOH, CAS 1310-73-2): dissolver 10 g em cerca de 70 mL de água e transferir para um balão volumétrico de 100 mL. Completar o volume e homogeneizar. Em um béquer, dissolver 2 g de acetato de chumbo nessa solução. Homogeneizar e transferir para um frasco âmbar com conta-gotas.
- Acetona (C3H6O, CAS 67-64-1)
- Ácido sulfúrico 98% (H2SO4, CAS 7664-93-9)
- Clorofórmio (CHCl3, CAS 67-66-3) ou tetracloreto de carbono (CCl4, CAS 56-23-5)
- Éter etílico (C4H10O, CAS 60-29-7) ou éter de petróleo
- Peróxido de hidrogênio (H2O2, CAS 7722-84-1)
- Solução de ácido acético 1:1 (v/v): em uma proveta de 100 mL, acrescentar 50 mL de água e, em seguida, acrescentar cuidadosamente ácido acético glacial (C2H4O2, CAS 64-19-7) até completar o volume de 100 mL. Homogeneizar e acondicionar a solução em frasco âmbar com conta-gotas.
- Solução de ácido clorídrico 1:1 (v/v): em uma proveta de 100 mL, acrescentar 50 mL de água e, em seguida, acrescentar cuidadosamente ácido clorídrico (HCl, CAS 7647-01-0) até completar o volume de 100 mL. Homogeneizar e acondicionar a solução em frasco âmbar com conta-gotas.
- Soluções de éter etílico ou éter de petróleo + clorofórmio ou tetracloreto de carbono preparadas nas proporções 10:90, 20:80, 30:70 e 40:60 (v/v)
- Solução estoque de Kastle-Meyer: pesar 1 g de fenolftaleína (C20H14O4, CAS 77-09-8) e 10 g de hidróxido de potássio (KOH, CAS 1310-58-3) e diluir em 50 mL de água. Adicionar 10 g de zinco em pó para que a solução fique transparente. Homogeneizar e acondicionar a solução em frasco âmbar sob refrigeração.
- Solução de Kastle-Meyer de trabalho: diluir 5 mL de solução estoque Kastle-Meyer em 20 mL de etanol (C2H5OH, CAS 64-17-5). Homogeneizar e acondicionar a solução em frasco âmbar com conta-gotas.
- Solução de molibdato de amônio 1% (m/v): pesar 1 g de molibdato de amônio tetrahidratado ((NH4)6Mo7O24.4H2O, CAS 12054-85-2) e dissolver em cerca de 50 mL de água, em um béquer. Transferir para balão volumétrico de 100 mL e completar o volume com água. Homogeneizar e acondicionar a solução em frasco âmbar com conta-gotas.
- Zinco em pó (Zn)
¶ C.1.5 Preparo das amostras
Avaliar a característica da amostra (peletizada, farelada, úmida e ingrediente de origem mineral) e, no caso de amostras fareladas, proceder diretamente conforme item “Preparo para análise microscópica”.
Nota: as amostras para análise microscópica não devem ser moídas.
¶ C.1.5.1 Amostras peletizadas
- Homogeneizar a amostra e transferir quantidade suficiente para um gral ou para uma bandeja metálica.
- Acrescentar água suficiente para umedecer a amostra, agitando até a completa desintegração dos pellets.
- Sob água corrente, fazer sucessivas lavagens da amostra sobre peneiras plásticas com aberturas de 0,1 mm a 0,2 mm (ou utilizar um saco de pano limpo) até o clareamento do filtrado, evitando a perda de partículas sólidas.
- Secar em estufa à temperatura de 50 °C a 70 °C.
- Fragmentar a amostra com pistilo até o desprendimento das partículas.
- Prosseguir conforme item “Preparo para análise microscópica” (C.1.6.1).
¶ C.1.5.2 Amostras úmidas
- Caso necessário, descongelar a amostra.
- Transferir a amostra para bandeja previamente identificada com os cinco primeiros dígitos do número de registro da amostra no laboratório.
- Secar em estufa à temperatura máxima de 70 °C.
- Peneirar para retirada dos itens mais grosseiros.
- Utilizar a fração mais fina para prosseguimento do preparo.
- Prosseguir conforme item “Preparo para análise microscópica”.
¶
¶ C.1.5.3 Amostras minerais
- Transferir a quantidade mínima de 15,00 g de amostra para uma tamis com malha de abertura adequada.
- Lavar com água corrente, desprezando a fase solúvel e transferir a fração retida para outro recipiente.
- Secar em estufa à temperatura máxima de 80 ºC.
- Tamisar cada uma das fases e transferir as porções obtidas para placas de Petri identificadas.
- Proceder à análise microscópica.
¶ C.1.6 Procedimento de análise
¶ C.1.6.1 Preparo para análise microscópica
O preparo da amostra para análise microscópica poderá ser executado utilizando copo de sedimentação ou funil de separação.
¶ C.1.6.1.1 Preparo em copo de sedimentação
- Homogeneizar a amostra e, caso necessário, fazer quarteamento.
- Pesar no mínimo 15,00 g de amostra em cálice ou cápsula com capacidade superior a 100 mL.
- Sob exaustão, adicionar 80 mL a 100 mL de clorofórmio ou tetracloreto de carbono, homogeneizar e aguardar a separação das fases.
- Retirar o sobrenadante com auxílio de espátula ou colher de cabo longo e transferir para outro recipiente, tomando cuidado para que o precipitado não escorra juntamente com o solvente.
- Recolher o precipitado em placa de Petri, papel filtro ou papel toalha para evaporação do excesso de solvente.
- No cálice com o sobrenadante e o solvente, adicionar éter até obter entre 10 e 30% deste na solução éter/clorofórmio, homogeneizar a amostra e aguardar a separação de fases.
- Retirar o sobrenadante com auxílio de espátula ou colher de cabo longo e transferir para placa de Petri, papel filtro ou papel toalha (essas etapas poderão ser repetidas adicionando progressivamente éter até obter o número de fases desejado).
- Descartar o solvente, utilizando uma peneira para conter o que restou do sobrenadante e transferir o precipitado para placa de Petri, papel filtro ou papel toalha.
- Secar todas as fases em estufa ou em capela de exaustão.
- Tamisar cada uma das fases e transferir as porções obtidas para placas de Petri identificadas.
- Proceder à análise microscópica.
¶ C.1.6.1.2 Preparo em funil de separação
- Homogeneizar a amostra e, caso necessário, fazer quarteamento.
- Pesar no mínimo 15,00 g de amostra e transferir para funil de separação.
- Sob exaustão, adicionar de 80 mL a 100 mL de clorofórmio ou tetracloreto de carbono, homogeneizar e aguardar a separação das fases.
- Abrir a torneira do funil para recolhimento do precipitado em uma cápsula. Se ocorrer entupimento no funil, utilizar uma haste metálica para auxiliar a drenagem do material. Tomar cuidado para que a fase sobrenadante não saia juntamente com o precipitado.
- Transferir o precipitado para placa de Petri, papel filtro ou papel toalha. Se necessário, utilizar funil de Büchner acoplado a um kitasato, drenando, sob vácuo, o excesso de solvente.
- No funil, adicionar à fração remanescente da amostra éter até obter entre 10 e 30% deste na solução éter/clorofórmio, homogeneizar e aguardar separação de fases.
- Retirar o precipitado em cápsula e transferir para placa de Petri, papel filtro ou papel toalha (essas etapas poderão ser repetidas adicionando progressivamente éter até obter o número de fases desejado).
- Descartar o solvente em bombona.
- Secar todas as fases em estufa ou em capela de exaustão.
- Tamisar cada uma das fases e transferir as porções obtidas para placas de Petri identificadas.
- Proceder à análise microscópica.
¶ C.1.6.2 Análise microscópica
- Proceder à tamisação da amostra com malhas de aberturas de no mínimo 1,4 mm, 1,0 mm e 0,5 mm, recolhendo o material sobre placas identificadas.
- Examinar ao microscópio estereoscópio cada uma das porções contidas nas placas de Petri, iniciando a análise a partir da fração mais grossa.
- Pesquisar de uma extremidade a outra da placa de Petri de maneira que todas as partículas sejam observadas em camada fina.
- Com auxílio de pinça, separar o fragmento e identificá-lo com base no seu tamanho, forma, cor, textura, dureza, brilho e outras características morfológicas, comparando-os com materiais de referência.
Nota: na indisponibilidade de padrões comerciais, podem ser utilizadas como materiais de referência amostras de subprodutos de origem animal (feed grade), oriundas de estabelecimentos fiscalizados pelo MAPA e previamente examinadas por analistas capacitados para a execução deste método.
¶ C.1.6.3 Testes qualitativos
A realização das provas qualitativas (tabela 1) é opcional e complementar ao exame microscópico. Retirar o fragmento suspeito com pinça pontiaguda e transferir para placa de Petri ou tubo de ensaio, adicionando os reagentes indicados na prova correspondente.
Nota: se necessárias, outras provas podem ser realizadas de acordo com a literatura relacionada. Recomenda-se consultar o "Guia Orientativo para Microscopia da Alimentação Animal" para provas adicionais.
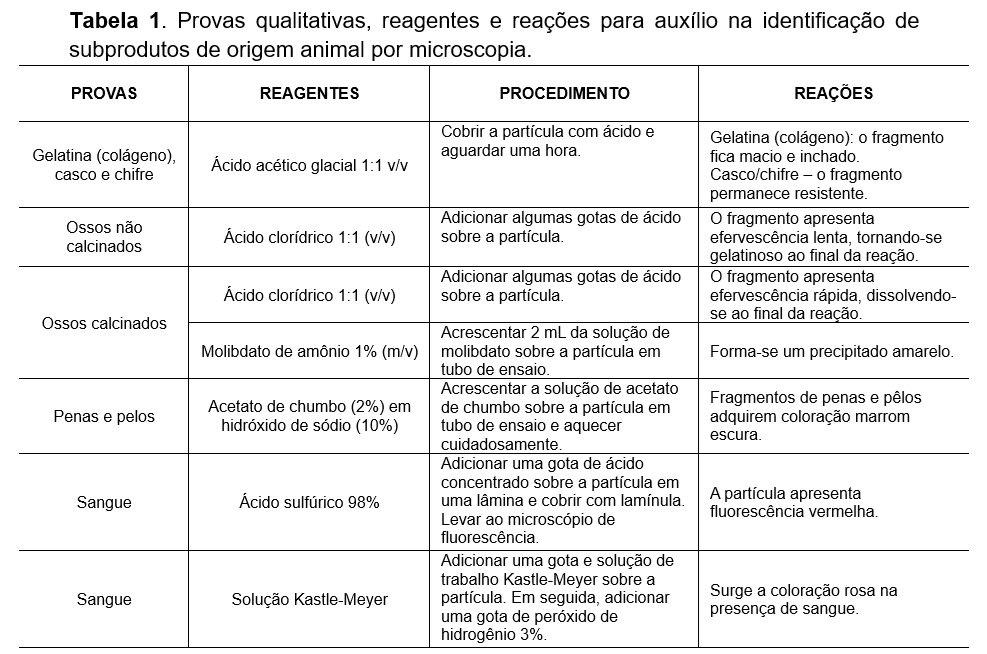
¶ C.1.7 Cálculos e expressão dos resultados
- Quando não são detectados subprodutos de origem animal, os resultados são expressos como “NÃO DETECTADO”.
- Quando são detectados subprodutos de origem animal, os resultados são expressos como “DETECTADO”, citando os achados microscópicos.
- Em ambos os casos, devem ser citados os limites de detecção dos analitos obtidos em procedimentos de validação intralaboratorial.
¶ Bibliografia
- American Association of Feed Microscopists (AAFM). Manual of microscopic analysis of feedstuffs. 3rd. ed., 1992 (p. 113 – 145).
- Brasil. Ministério da Agricultura e Pecuária. Secretaria de Defesa Agropecuária. Guia Orientativo para Microscopia da Alimentação Animal. Disponível em: https://wikisda.agricultura.gov.br/pt-br/Laborat%C3%B3rios/Metodologia/ALA/Guia_microscopia_ALA.
- Huss, W. Microscopy and quality control in the manufature of animals feed. Hohenheim: Roche, 1975, 63 p. (Information service).
- Khajarern, J.; Khajarern, S. Manual of feed microscopy and quality control. American Soybean Association. Khon Kaen (Tailândia): 1999 (3ª edição), 256 p.
¶ 5. Base legal e documentos de referência
¶ 5.1 Base legal
- Brasil. Decreto nº 12.031, de 28 de maio de 2024. Regulamenta a Lei nº 6.198, de 26 de dezembro de 1974, e a Lei nº 14.515, de 29 de dezembro de 2022, para dispor sobre a inspeção e a fiscalização obrigatórias dos produtos destinados à alimentação animal. Brasília: Diário Oficial da União, seção 1, p. 7 (29 de maio de 2024).
- Brasil. Ministério da Agricultura, Pecuária e Abastecimento. Instrução Normativa nº 15, de 26 de maio de 2009. Regulamenta o registro dos estabelecimentos e dos produtos destinados à alimentação animal. Brasília: Diário Oficial da União, seção 1 (28 de maio de 2009).
- Brasil. Ministério da Agricultura e Pecuária. Portaria nº 747, de 23 de dezembro de 2024. Estabelece os critérios e requisitos para o credenciamento e fiscalização de laboratórios pelo Ministério da Agricultura e Pecuária. Brasília: Diário Oficial da União, seção 1, p. 8 (27 de dezembro de 2024).
¶ 5.2 Documentos de referência
- Associação Brasileira de Normas Técnicas. Norma brasileira ABNT NBR 5891. Regras de arredondamento na numeração decimal. Rio de Janeiro: ABNT, 2015. ISBN: 978-85-07-05281-4.
- Food and Agriculture Organization of the United Nations (FAO). Quality assurance for animal feed analysis laboratories. FAO Animal Production and Health Manual No. 14. Rome: FAO, 2011. ISBN: 978-92-5-107050-5.
- União Europeia. Regulamento de execução (UE) 2024/771 da Comissão de 29 de fevereiro de 2024 que altera o Regulamento (CE) n.° 152/2009 que estabelece os métodos de amostragem e análise para o controlo oficial dos alimentos para animais. Bruxelas: Jornal Oficial da União Europeia (15 de março de 2024). ISSN: 1977-0774.
¶ 6. Disposições gerais
As sugestões para aprimoramento ou possíveis correções deste documento devem ser direcionadas ao Departamento responsável, para alinhamento das melhores práticas de mercado, legislação vigente e/ou regulamentações, que não tenham sido contempladas na versão vigente.
¶ 7. Histórico de revisões
| Versão | Conteúdo alterado | Data | Motivo |
|---|---|---|---|
| 1 | - | 15/04/2025 | Elaboração do documento. |
| 2 | B.7.6 | 22/07/2025 | Correção do título (inclusão do analito fósforo). |
| 3 | B.3 | 25/11/2025 | Correção do método de referência. |
| 3 | B.6 | 25/11/2025 | Correção da unidade de expressão do resultado. |
| 3 | B.7.2 | 25/11/2025 | Inserido método 2021.004 como alternativa. |
| 3 | B.7.4 | 25/11/2025 | Inserido item B.7.4.1 para cálculo da relação P/F. Menções ao cálculo nos itens B.7.3 e B.7.4. |
| 4 | 4.A | 22/01/2026 | Excluída necessidade de refrigeração de amostras de farinhas, gorduras e óleos até o preparo para análise. |
| 5 | B.9.1.5 | 28/01/2026 | Alterado critério de aceitação para recuperação de nitrogênio. Excluída necessidade de ser MRC. |
| 6 | B.7.7.4 | 12/02/2026 | Correção do preparo da solução de ácido nítrico 1:4 (v/v). |
| 7 | B.7.3.1 | 11/03/2026 | Inclusão/descrição do método para flúor com extração assistida por ultrassom (Doc. SEI 5104155). |
| 7 | B.8.1 | 11/03/2026 | Inclusão/descrição do método para matéria mineral (Doc. SEI 5104155). |