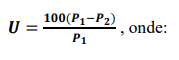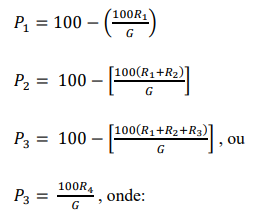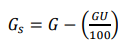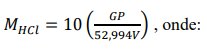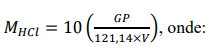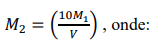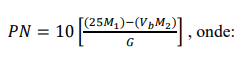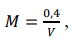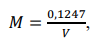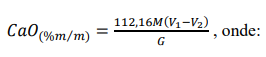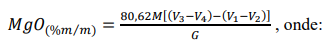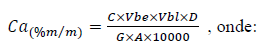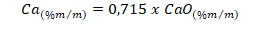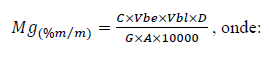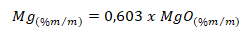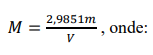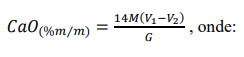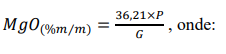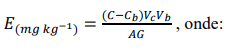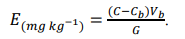Elaboração, distribuição, informações: Ministério da Agricultura e Pecuária Secretaria de Defesa Agropecuária - SDA Departamento de Serviços Técnicos - DTEC Esplanada dos Ministérios, Bloco D, Anexo, Ala B, 4º andar, sala 433 CEP: 70043-900, Brasília - DF www.agricultura.gov.br e- mail: cgal@agro.gov.br Central de Relacionamento: 0800 704 1995
Reunir os métodos analíticos a serem utilizados na verificação da conformidade dos insumos agrícolas quanto aos teores de nutrientes e quanto à presença de contaminantes químicos, nas análises realizadas para fiscalização destes produtos pelos laboratórios oficiais ou credenciados do Ministério da Agricultura e Pecuária (Mapa).
Processo:
Análises Laboratoriais
Entrega:
Segurança e qualidade de insumos agropecuários
Público alvo e demais interessados:
Laboratórios oficiais ou credenciados do Ministério da Agricultura e Pecuária (Mapa)
Versão do documento:
2
Setor responsável e responsabilidades
A Coordenação Geral de Laboratórios Agropecuários do Departamento de Serviços Técnicos é responsável pela elaboração, atualização e envio para aprovação deste manual, tendo responsabilidade quanto aos procedimentos descritos no documento.
O presente manual possui vigência e prazo indeterminado e será revisado sempre que necessário, no mínimo anualmente, pela Coordenação Geral de Laboratórios Agropecuários do Departamento de Serviços Técnicos (CGAL/DTEC).
A gestão desse manual está sob a responsabilidade da CGAL/DTEC que prestará auxílio ao público alvo leitor dúvidas e/ou sugestões quanto à aplicação deste manual devem ser submetidas ao Departamento responsável.
A publicação e atualização das versões na plataforma oficial da SDA para acesso pelo público alvo será de responsabilidade da Secretaria representada pelo DTEC.
O objetivo do Manual é o de reunir os métodos analíticos a serem utilizados na verificação da conformidade dos insumos agrícolas quanto aos teores de nutrientes e quanto à presença de contaminantes químicos, nas análises realizadas para fiscalização destes produtos pelos laboratórios oficiais ou credenciados do Ministério da Agricultura e Pecuária (Mapa). Seu uso em laboratórios de controle de qualidade de empresas produtoras, laboratórios privados e outros é facultativo. Outros métodos podem ser empregados nas análises de controle de qualidade realizadas por estes laboratórios, desde que sejam comprovadamente equivalentes e validados, quando a finalidade é de comparação com os resultados obtidos pelos métodos oficiais.
O Ministério da Agricultura e Pecuária apresentou métodos oficiais para o controle de qualidade dos fertilizantes, corretivos de acidez de solos e inoculantes pela primeira vez em 1983 (Brasil, 1983), a partir do trabalho do Professor Doutor José Carlos Alcarde, da Escola Superior de Agricultura Luiz de Queiróz / Universidade de São Paulo. Este compêndio de métodos foi reeditado em 1988.
Em 2007 foi oficializada, por intermédio da Instrução Normativa SDA nº 28, de 27 de junho de 2007, a primeira revisão e ampliação do compêndio original de métodos. A segunda revisão e ampliação foi oficializada pela Instrução Normativa SDA nº 03, de 26 de janeiro de 2015. A terceira veio a público por meio da Instrução Normativa nº 37, de 13 de outubro de 2017. Este trabalho representa a quarta revisão, ampliação e atualização do Manual de Métodos.
O objetivo do Manual é o de reunir os métodos analíticos a serem utilizados na verificação da conformidade dos insumos agrícolas quanto aos teores de nutrientes e quanto à presença de contaminantes químicos, nas análises realizadas para fiscalização destes produtos pelos laboratórios oficiais ou credenciados do Ministério da Agricultura e Pecuária (Mapa). Seu uso em laboratórios de controle de qualidade de empresas produtoras, laboratórios privados e outros é facultativo. Outros métodos podem ser empregados nas análises de controle de qualidade realizadas por estes laboratórios, desde que sejam comprovadamente equivalentes e validados, quando a finalidade é de comparação com os resultados obtidos pelos métodos oficiais.
De modo geral, estão apresentados métodos internacionalmente aceitos, alguns deles métodos clássicos da Química Analítica, propostos com o suporte da instrumentação necessária e na medida da precisão e exatidão exigida pela legislação brasileira. São basicamente os mesmos métodos empregados por outras entidades reguladoras e fiscalizadoras ao redor do mundo, que os empregam com o mesmo objetivo, como poderá ser verificado nas referências bibliográficas enumeradas ao final. Estão divididos em temas, aplicados de acordo com a classificação e composição dos insumos, a saber:
I. Análise de fertilizantes minerais destinados à aplicação via solo.
II. Análise de fertilizantes minerais destinados à aplicação foliar, cultivo hidropônico, fertirrigação, aplicação via semente e das soluções para pronto uso.
III. Análise dos fertilizantes orgânicos, organominerais e biofertilizantes destinados à aplicação via solo.
IV. Análise dos fertilizantes orgânicos, organominerais e biofertilizantes destinados à aplicação foliar, cultivo hidropônico, fertirrigação, aplicação via semente e das soluções para pronto uso.
V. Análise dos corretivos de acidez.
VI. Análise de substratos e condicionadores de solo.
VII. Análise de remineralizadores de solo.
VIII. Métodos complementares.
Alguns cuidados operacionais e recomendações de trabalho não foram repetidos de forma sistemática no texto dos métodos analíticos, pois estão compreendidos nas BOAS PRÁTICAS ANALÍTICAS as quais são requisitos indispensáveis para o correto desenvolvimento dos trabalhos e devem sempre ser atendidas. A seguir são listados alguns pontos essenciais que devem ser observados, alguns deles visando dar flexibilidade ao trabalho do analista, sem prejuízo da precisão, exatidão e consistência de seu trabalho e confiabilidade de seus resultados:
a) Uso de material de proteção individual: Neste Manual não é feita referência ao uso de equipamentos de proteção individual (EPIs) embora o seu uso seja fortemente encorajado. Luvas adequadas, jalecos, sapatos fechados e óculos de segurança devem ser empregados durante a realização de todas as atividades no laboratório. Manipular ácidos concentrados e produtos voláteis de qualquer natureza somente em capela. Manter uma disposição de permanente atenção e cuidado com a operação que se está desenvolvendo.
b) Qualidade da água a utilizar: A qualidade mínima da água a ser empregada será a da água deionizada ou destilada. Toda e qualquer referência simples a “água” nas descrições dos métodos pressupõe esta pureza mínima. Em casos especiais, que serão indicados no método de análise, esta água deverá ser submetida a processos específicos de purificação, que implicam melhor qualidade. Caso o laboratório tenha recursos disponíveis, água de qualidade superior à explicitada acima pode ser empregada em todas as operações.
Nas operações de dissolução, diluição, enxágue ou lavagens mencionadas nos métodos de análise, sem especificação da natureza dos solventes ou diluentes, está implícita a utilização de água.
c) Material empregado nos métodos analíticos: O material corrente de laboratório não está especificado quando da descrição dos métodos, salvo quanto à sua capacidade. A descrição dos itens incluída nos métodos analíticos limita-se a aparelhos e utensílios especiais ou àqueles que requerem exigências específicas. Relativamente ao material de vidro graduado, o laboratório deverá assegurar-se de seu grau de precisão, tomando como referência as normas metrológicas apropriadas.
d) Limpeza do material: O material deve estar bastante limpo, podendo requerer uma limpeza especial (que nestes casos será descrita), sobretudo quando as determinações incidem sobre teores muito baixos do elemento a analisar.
e) Qualidade dos reagentes: Salvo disposições contrárias claramente mencionadas nos métodos de análise, todos os reagentes deverão ser de pureza analítica (p.a.). Em casos específicos, que serão igualmente ressaltados, poderá ser exigida uma pureza maior.
f) Calibração e manutenção de equipamentos e vidraria: Os laboratórios poderão definir prazos e políticas próprias de manutenção e calibração de equipamentos e vidraria, atentando às regras previstas em seu sistema de controle da qualidade e na Norma adotada.
g) Medidas de massa, volume, tempo e temperatura: Para os métodos analíticos apresentados neste Manual por vezes serão solicitadas medidas que não necessitam ter, rigorosamente, o valor expresso, exceto quando especificado no método, como, por exemplo, em casos de padronização, preparação de soluções de referência e outros. Na ausência de uma referência clara a uma pesagem exata ou ao uso de vidraria volumétrica específica, não é necessário usar equipamento de maior precisão do que a solicitada e vidraria de volumes próximos pode ser utilizada conforme a disponibilidade e conveniência do laboratório. Desta forma, também é permitido flexibilizar as massas pesadas das amostras, as concentrações de soluções padronizadas, o volume dos balões utilizados nas curvas de calibração e soluções de leitura das amostras, de acordo com o teor especificado ou esperado, desde que registrado o valor exato para uso nos cálculos finais.
h) Procedimentos de extração: Em algumas análises, a extração é empírica e poderá não ser quantitativa, dependendo do produto e seus diversos componentes. Por exemplo, no caso de alguns óxidos de manganês a quantidade extraída (extração ácida) poderá não traduzir a quantidade total de manganês do produto. Cabe ao fabricante providenciar para que o teor declarado corresponda de fato à quantidade extraída nas condições previstas no método. Assim, em algumas situações, o chamado teor “total” corresponde, na verdade, ao teor extraído nas condições enérgicas descritas pelo método.
i) Uso de materiais de referência e amostras de controle. Participação em Programas de Ensaios de Proficiência: O emprego de compostos químicos padrões, estáveis e de composição bem definida, de amostras-controle (amostras com teores conhecidos, mas obtidas sem processos formais de certificação) e de materiais de referência certificados (MRC) deve ser uma prática rotineira do laboratório, para verificar o funcionamento dos equipamentos e a execução correta das técnicas analíticas. As amostras de controle podem ser preparadas no próprio laboratório, a partir de amostras homogêneas, analisadas repetidas vezes para obter uma estimativa razoável dos valores verdadeiros e dos intervalos de confiança para os resultados dos elementos ou índices desejados. Seu uso, bem como o de materiais de referência certificados, possibilita a avaliação da conformidade das atividades de rotina e a consequente garantia da qualidade dos resultados obtidos. A participação em Programas de Ensaios de Proficiência será igualmente uma atividade fundamental para a garantia da qualidade dos trabalhos executados, buscando monitorar as diferentes situações de procedimentos aplicados a diferentes matrizes. Com relação à secagem e armazenamento dos materiais de referência certificados (MRC), se o certificado informar condições diferentes das que são descritas neste Manual, seguir o indicado pelo fabricante do material.
j) Preparo de curvas de calibração: As curvas de calibração recomendadas neste Manual são sugestões, podendo sofrer alterações conforme as características de cada equipamento empregado. Podem-se tomar soluções-padrão de concentrações diferentes, desde que obedecidas as faixas lineares de trabalho, bem como variar o número de pontos na curva, desde que se empregue o mínimo de três pontos mais o “zero” (quando este faz parte da curva de calibração; caso não faça parte, devem ser usados no mínimo quatro padrões para a construção da curva), e desde que o princípio do método analítico empregado não seja alterado. Observar com cautela as alterações feitas de modo a manter o padrão no ambiente químico relativo ao método empregado e que o pH e a viscosidade da solução não interfiram nas determinações em métodos espectrofotométricos ou instrumentais.
k) Uso de soluções padrão multielementares: o uso de padrões multielementares, especialmente para as determinações por espectrometria de absorção atômica, é permitido, devendo ser ressalvados os casos de interferências.
l) Tratamento de resíduos de laboratório: de modo geral, não se faz referência à separação e destinação dos resíduos gerados. Porém nenhum resíduo do laboratório químico deve ser descartado no esgoto normal ou no ambiente sem prévia avaliação e definição da forma de disposição e tratamento adequados.
A par de toda a evolução técnico-instrumental e dos recursos disponibilizados aos laboratórios cabe lembrar que o principal agente do trabalho analítico é o técnico responsável pela sua execução. É fundamental a sua capacitação, habilidade e atitude profissional, que devem ser priorizadas e valorizadas na medida de sua relevância.
¶ 4. Procedimentos - ANÁLISE DE CORRETIVOS DE ACIDEZ
Homogeneizar a amostra e dividi-la, por quarteação, em duas frações: uma destinada à análise granulométrica e a outra às análises químicas.
A parte da amostra que será destinada à análise granulométrica deverá ser reduzida por quarteação cuidadosa a uma massa entre 100 e 150 gramas, que deverá ser previamente secada em estufa à temperatura de 105 ± 5ºC, até peso constante.
NOTA 206: Para amostras com teor de umidade tal que justifique a execução da análise granulométrica por via úmida, como descrito à frente no item B.2.b, a parte da amostra reservada à análise granulométrica não deverá ser secada.
A fração destinada às análises químicas deverá ser reduzida por quarteação cuidadosa a aproximadamente 60 gramas. Pesar e registrar a massa da amostra in natura (P1). Transferir para vidro de relógio ou cápsula de porcelana previamente tarados e levar à secagem em estufa a 105 ± 5 ºC até peso constante. Retirar, deixar esfriar em dessecador e, após esfriar, pesar o conjunto e determinar a massa da amostra secada (P2).
Estes dados serão utilizados no cálculo da umidade (U), sendo:
P1: massa da amostra in natura, em gramas.
P2: massa da amostra, após a secagem, em gramas.
Esta massa da amostra secada deverá ser totalmente moída e passada em peneira com abertura de malha de 300 µm e destinada às análises químicas do Poder de Neutralização (PN), Óxido de Cálcio (CaO) e Óxido de Magnésio (MgO).
Amostras coletadas com massa menor que 100 gramas deverão ter sua análise cancelada. Para aquelas com massa entre 100 e 200 gramas, executar apenas as análises químicas.
NOTA 207: Caso a amostra de corretivo aumente a sua massa após ser seca na estufa, ou seja, a massa da amostra, após a secagem, seja maior que a massa da amostra in natura, a amostra seca em estufa deve ser descartada e as análises granulométrica e químicas de corretivo devem ser realizadas somente com a amostra in natura.
a) Pesar a fração da amostra reservada para a análise granulométrica, com precisão de 0,01 g.
b) Transferir sobre as peneiras encaixadas uma sobre a outra, em ordem crescente de abertura das malhas, ficando a de maior abertura de malha acima e o recipiente de fundo abaixo da última peneira.
c) Agitar durante 10 minutos, no agitador mecânico. Pesar cada peneira mais os retidos com aproximação de 0,01 g e calcular as frações retidas em cada uma.
d) Calcular a porcentagem de massa passante em cada peneira, de acordo com as expressões:
P1: porcentagem em massa passante na peneira com abertura de 2,00 mm.
P2: porcentagem em massa passante na peneira com abertura de 840 µm.
P3: porcentagem em massa passante na peneira com abertura de 300 µm.
G = massa da amostra, em gramas.
R1 = massa do material retido na peneira de 2,00 mm, em gramas.
R2 = massa do material retido na peneira de 840 µm, em gramas.
R3 = massa do material retido na peneira de 300 µm, em gramas.
R4 = massa do material recolhido no recipiente de fundo, em gramas.
Aplicável aos corretivos que apresentem teor de umidade que impossibilite a realização da análise granulométrica pelo procedimento usual, descrito anteriormente. Esta condição deverá ser informada pelo produtor e verificada.
a) Pesar a fração da amostra reservada para tal, com precisão de 0,01 g.
b) Transferir para as peneiras, como no procedimento anterior.
c) Lavar com um fluxo moderado de água de torneira, até que a água que passa através das peneiras esteja límpida. Tomar cuidado para evitar perda da amostra por respingos.
d) Secar as peneiras com os retidos a 65 ± 5 ºC, até peso constante. Esfriar, pesar cada peneira mais o retido com aproximação de 0,01 g e calcular a fração retida nas peneiras.
e) Calcular a porcentagem de massa passante em cada peneira de acordo com as expressões anteriormente descritas, usando nas fórmulas de cálculo o valor referente à massa seca (Gs), descontada a umidade, sendo:
onde U é a porcentagem de umidade da amostra e G a massa da amostra in natura tomada para a análise granulométrica, em gramas.
Justificativa: como a amostra in natura apresenta teor significativo de umidade, deve-se considerar nos cálculos a massa da amostra em base seca. Caso contrário, a massa de água relativa ao teor de umidade irá somar-se à fração passante pelas peneiras. Portanto, nas fórmulas de cálculo, a massa da amostra (G) deverá ser substituída por Gs.
NOTA 208: Os procedimentos de análise granulométrica se aplicam também aos corretivos de alcalinidade e sodicidade que se apresentem em pó, assim como o produto sulfato de cálcio, quando registrado como condicionador de solo.
Os corretivos de acidez, alcalinidade e sodicidade especificados como “ultrafino” ou “filler” deverão ser avaliados com relação ao percentual passante na peneira de 300 micrometros (0,30 mm).
Verificando-se especificação em peneira(s) com abertura de malha não referida, promover a análise segundo o processo descrito, utilizando-se as peneiras com abertura de malha especificadas pelo fabricante e constantes do registro do produto.
Fundamenta-se em colocar em contato uma massa conhecida do corretivo de acidez com uma quantidade conhecida e em excesso de solução de ácido clorídrico padronizada, fazendo com que o corretivo neutralize uma parte do ácido. O excesso de ácido será quantificado por alcalimetria, obtendo-se indiretamente quanto do ácido foi neutralizado pelo corretivo, por titulação com indicador ou com indicação potenciométrica. Aplicável aos corretivos de acidez do solo.
i. Solução alcoólica de fenolftaleína a 1 % (m/v): pesar 1 g do indicador fenolftaleína p.a. e diluir a 100 mL com álcool etílico p.a
ii. Solução do indicador alaranjado de metila a 0,1 % (m/v): dissolver 0,1 g do indicador alaranjado de metila p.a. em água e completar o volume a 100 mL.
iii. Solução de HCl 0,5 ± 0,01 mol L-1, padronizada: diluir 42 mL de HCl concentrado p.a. em água, transferir para balão volumétrico de 1 litro, completar o volume e homogeneizar.
iv. Carbonato de sódio (Na2CO3), p.a., padrão primário, previamente secado por 2h a 280-290 ºC em forno elétrico, ou seguindo-se a recomendação do fabricante/produtor quanto à secagem do material, e conservado em dessecador. Como alternativa, pode ser utilizado o padrão primário tris-hidroximetil amino metano (TRIS, massa molar 121,14 g/mol), também secado a 110 ± 10 ºC até peso constante e conservado em dessecador ou seguindo-se a recomendação do fabricante/produtor quanto a secagem do material.
v. Solução de vermelho de metila em etanol: dissolver 0,2 g de vermelho de metila, p.a., em 60 mL de etanol, p.a., e diluir com água a 100 mL.
Padronização da solução de HCl 0,5 mol L-1:
i. Com Na2CO3:
a) Pesar uma massa (G) de 0,5 g de Na2CO3 com precisão de 0,1 mg e transferir para erlenmeyer de 250 – 300 mL. Adicionar 50 – 70 mL de água, agitar com cuidado até a completa dissolução do sal e adicionar 4 a 5 gotas da solução de alaranjado de metila.
b) Transferir a solução de HCl para uma bureta de 25 ou 50 mL e titular a solução do erlenmeyer até esta começar a apresentar variação de cor.
c) Ferver suavemente a solução do erlenmeyer por 2 minutos (para eliminação do CO2), esfriar em água corrente até a temperatura ambiente e prosseguir a titulação até a solução apresentar a coloração levemente avermelhada, diferenciada da coloração de uma solução de referência preparada com 80 mL de água fervida e a mesma quantidade em gotas do indicador.
d) Anotar o volume gasto. Repetir mais duas vezes, calcular as concentrações e fazer a média dos valores encontrados.
e) O cálculo da concentração da solução ácida é dado pela expressão:
G: massa exata de Na2CO3 que foi pesada em cada evento.
V: volume da solução de HCl gasto na titulação, em mililitros.
P: porcentagem de pureza do reagente padrão (Na2CO3) utilizado.
ii. com tris-hidroximetil-amino-metano (TRIS):
a) Pesar uma massa (G) de 0,5 g de TRIS com precisão de 0,1 mg e transferir para erlenmeyer de 125 mL. Adicionar 20 mL de água, agitar com cuidado até a completa dissolução do reagente e adicionar 4 a 5 gotas da solução de alaranjado de metila.
b) Titular a solução do erlenmeyer até começar a apresentar variação de cor (ponto de viragem do amarelo para laranja);
c) Anotar o volume gasto (V). Repetir mais duas vezes, calcular a concentração pela expressão abaixo, utilizando as massas pesadas de TRIS. Fazer a média das concentrações encontradas.
M = concentração da solução ácida em mol L-1;
V = volume da solução ácida gasto na titulação, em mililitros;
P = pureza do reagente padrão utilizado, em porcentagem em massa;
G = massa exata de TRIS que foi pesada, em gramas.
NOTA 209:
i. Soluções padrões de HCl também podem ser obtidas a partir de soluções padrões de qualidade referenciada, adquiridas prontas. De qualquer forma, devem ser padronizadas periodicamente.
ii. A concentração final da solução de HCl poderá variar na faixa de 0,5 ±0,01 mol L-1. Se diferir deste valor, corrigir por concentração com ácido clorídrico (1+1) ou por diluição com água.
d) Solução de NaOH 0,25 mol L-1, padronizada. Pesar 10 gramas do reagente (NaOH), p.a. e dissolver em água. Transferir para balão volumétrico de 1 litro, completar o volume e homogeneizar.
Padronização:
a) Tomar 10 mL da solução de HCl 0,5 mol L-1 padronizada e transferir para erlenmeyer de 250 mL.
b) Fazer um volume de aproximadamente 50 mL com água e acrescentar 3-5 gotas da solução alcoólica de fenolftaleína.
c) Titular com a solução de NaOH aproximadamente 0,25 mol L-1 até verificar-se a viragem de incolor para uma leve cor rosada do indicador.
d) Repetir mais duas vezes e fazer a média das concentrações obtidas.
A concentração M2 deve ser calculada por:
M1 = concentração exata da solução de HCl padronizada, em mol L-1.
V = volume gasto no procedimento de titulação, em mililitros.
a) Pesar, com precisão de 0,1 mg, uma massa da amostra de 1 g, se calcário, ou 0,5 g, se calcário calcinado, cal virgem, cal hidratada ou óxidos de cálcio e magnésio. Deve-se tomar a parte da amostra que foi secada, moída e passada em peneira de 0,30 mm. Esta massa da amostra será identificada nos cálculos como “G”.
b) Transferir para erlenmeyer de 250 mL, adicionar exatamente 50 mL da solução de HCl 0,5 mol L-1 padronizada, cobrir com vidro de relógio e ferver suavemente por 5 minutos. Esfriar, transferir para balão de 100 mL e completar o volume com água. Homogeneizar bem e filtrar em papel de filtro de porosidade média para um recipiente seco. Cuidado: após filtrar, não lavar o retido no papel de filtro. Esta solução é o extrato-amostra e será utilizada, também, na determinação dos teores de cálcio (como CaO) e magnésio (como MgO).
c) Pipetar 50 mL e transferir para erlenmeyer de 125 mL.
d) Acrescentar 3-5 gotas da solução de fenolftaleina e titular o excesso do ácido com a solução padronizada de NaOH 0,25 mol L-1, até o aparecimento de uma leve cor rosada do indicador. Anotar o volume gasto (Vb).
NOTA 210: Caso a solução a ser titulada (extrato-amostra) fique rosa sem a adição de NaOH ou com a adição de poucas gotas de NaOH, deve-se repetir o procedimento de extração com a metade da massa utilizada originalmente.
e) Calcular o poder de neutralização (PN) do corretivo, em porcentagem em massa de CaCO3 equivalente, pela expressão:
M1 = concentração da solução de HCl, em mol L-1.
Vb = volume (mL) da solução de NaOH gasto na titulação.
Para produtos escuros, para os quais há dificuldade de visualização do ponto final da titulação com o uso do indicador fenolftaleína, esta poderá ser efetuada tendo o ponto final de neutralização do excesso de HCl indicado potenciometricamente, ao se atingir o pH 7:
a) Preparar o extrato-amostra da mesma forma descrita acima em 4.C.1.4.a e 4.C.1.4.b.
b) Pipetar 50 mL e transferir para um béquer de 150 mL.
c) Colocar o béquer sobre o agitador magnético, introduzir o eletrodo de pH na solução e posicionar a bureta contendo a solução padronizada de NaOH, para a titulação.
d) Acionar o agitador magnético, promovendo uma agitação moderada com o magneto e titular cuidadosamente com a solução padronizada de NaOH até o pH atingir o valor 5.
e) Continuar a titulação gota a gota até o pH atingir o valor 7 e assim permanecer por um minuto, mantendo-se a agitação. Anotar o volume gasto (Vb).
f) Proceder ao cálculo do PN (poder de neutralização) da mesma forma descrita em 4.C.1.4.e.
¶ 2. ÓXIDO DE CÁLCIO E ÓXIDO DE MAGNÉSIO – Método complexométrico do EDTA
O método fundamenta-se na solubilização do cálcio e magnésio contidos no corretivo em meio ácido e sua determinação por volumetria com EDTA. Os resultados são apresentados como porcentagem em massa de seus óxidos. Aplicável a corretivos com teor de CaO ≥ 7% e MgO ≥ 8%. Não aplicável a produtos com alto teor de fosfato, ferro, manganês, zinco e outras impurezas (Mn e Zn ≤ 0,25% ).
a) Solução de ácido clorídrico, HCl, (1+1), com água.
b) Solução de ácido nítrico, HNO3, (1+1), com água.
c) Solução de hidróxido de potássio-cianeto de potássio: dissolver 280 g de KOH e 2 g de KCN (Cuidado: VENENO!), em 1 litro de água. Usar reagentes p.a.
d) Indicadores: calceína ou calcon:
- Calceína - opção 1: moer a mistura formada de 0,2 g de calceína, 0,12 g de timolftaleina e 20 g de nitrato de potássio (KNO3). Passar em peneira de abertura de 0,50 mm (500 µm). Homogeneizar bem. Viragem: verde para vinho (sem reflexos de verde). Usar reagentes p.a.
- Calceína – opção 2: juntar 0,1 g de calceína e 10 g de cloreto de sódio (NaCl), homogeneizar bem e moer, passando em peneira de abertura de 0,50 mm (500 µm). A viragem é de verde para laranja (isento de reflexos verdes). Usar reagentes p.a.
- Calcon (ácido calconcarbônico): transferir 0,100 g de calcon para um béquer de 100 mL, contendo 10 mL de trietanolamina e 10 mL de álcool metílico – reagentes p.a. Homogeneizar, esperar dissolver, transferir para recipiente de plástico e conservar em geladeira (duração: 30-45 dias). Viragem: vermelho para azul.
e) Solução padrão de CaCO3 0,020 mol L-1: dissolver 2,0000 g de carbonato de cálcio, CaCO3 padrão primário, previamente secado a 105 ºC - 110 ºC por 1 hora, ou seguindo-se a recomendação do fabricante/produtor quanto a secagem do material, em um volume mínimo de solução de HCl (1+1) e completar o volume a 1 litro, com água. Alternativamente pode ser utilizada solução de cálcio de 1000 mg L-1, adquirida pronta para o uso, com rastreabilidade e grau de pureza analítica adequados.
f) EDTA - sal dissódico di-hidratado do ácido etilenodiamino tetracético (C10H14N2Na2O8. 2H2O), p.a., previamente secado a 70-80ºC, por 2 horas, ou seguindo-se a recomendação do fabricante/produtor quanto a secagem do material.
g) Solução de EDTA 0,020 mol L-1: dissolver 7,4450 g de EDTA em água, completar o volume a 1 litro e homogeneizar.
Padronização da Solução deEDTA 0,020 mol L-1:
i. Transferir 20 mL da solução de CaCO3 0,020 mol L-1 para erlenmeyer de 250 mL ou 5 mL da solução padrão de Ca pronta para uso.
ii. Adicionar 50 mL de água aproximadamente, 5 mL da solução KOH-KCN e uma porção (15±1mg) do indicador calceína, ou 6 gotas da solução do indicador calcon, agitando após a adição de cada reagente.
iii. Titular imediatamente o cálcio com a solução de EDTA 0,020 mol L-1, agitando continuamente até a mudança permanente da cor do indicador: a calceína muda de verde fluorescente para roxo; o calcon muda de vinho para azul puro. Anotar o volume da solução de EDTA consumido. Repetir por mais duas vezes e fazer a média das concentrações encontradas. A concentração da solução de EDTA, em mol L-1, será dada por:
usando-se a solução de CaCO3 0,020 mol L-1 , ou
usando-se a solução padrão de Ca de 1000 mg L-1 , onde
V = é o volume da solução de EDTA gasto na titulação.
h) Solução tampão de pH 10: dissolver 67,5 g de cloreto de amônio (NH4Cl) em água, acrescentar 570 mL de hidróxido de amônio (NH4OH) concentrado, 2 g de cianeto de potássio (KCN – Cuidado: VENENO!), 50 mL de trietanolamina e 0,616 g de sulfato de magnésio (MgSO4.7H2O) e 0,931 g de EDTA dissódico di-hidratado. Completar o volume a 1 litro e homogeneizar. Usar reagentes p.a.
i) Solução de negro de eriocromo T a 0.5 % m/v: dissolver 0,25 g do indicador e 2 g de cloridrato de hidroxilamina em 50 mL de metanol. Estável por 20-25 dias; conservar em geladeira. Usar reagentes p.a.
j) Solução de KCN a 2%em massa/volume: dissolver 2 g de KCN p.a. em 100 mL de água (Cuidado: VENENO!).
Utilizar o extrato-amostra preparado para a determinação do poder de neutralização (PN) obtido em 4.C.1.4.b.
¶ 2.4. Determinação e cálculo do teor de óxido de cálcio (CaO)
a) Transferir 5 mL do extrato para erlenmeyer de 125 mL.
b) Adicionar 50 mL de água aproximadamente, 5 mL da solução KOH-KCN, duas gotas da solução de trietanolamina (1+1) e uma porção (15± 1) mg do indicador calceína, ou 6-8 gotas da solução do indicador calcon, agitando após a adição de cada reagente.
c) Titular imediatamente o cálcio com a solução de EDTA 0,020 mol L-1, agitando continuamente até a mudança permanente da cor do indicador: a calceína muda de verde fluorescente para roxo; o calcon muda de vinho para azul puro. Anotar o volume (V1) da solução de EDTA consumido.
d) Desenvolver, em paralelo, uma prova em branco e anotar o volume consumido (V2).
e) Calcular a porcentagem em massa de CaO, pela expressão:
M = concentração da solução de EDTA, em mol L-1.
V1 = volume (mL) da solução de EDTA padronizado gasto na titulação.
V2 = volume (mL) da solução de EDTA padronizado gasto na titulação da prova em branco.
G = massa inicial da amostra, em gramas.
¶ 2.5. Determinação e cálculo do teor de óxido de magnésio (MgO)
a) Transferir 5 mL do extrato para erlenmeyer de 125 mL.
b) Adicionar 50 mL de água aproximadamente, 5 mL da solução tampão de pH 10, 2 mL da solução de KCN a 2%, duas gotas da solução de trietanolamina (1+1) e 6-8 gotas da solução de negro de eriocromo T, agitando após a adição de cada reagente.
c) Titular imediatamente o cálcio mais magnésio com a solução de EDTA 0,020 mol L-1padronizada até a viragem do indicador, da cor vermelha vinho para azul puro e estável. Anotar o volume (V3) da solução de EDTA consumido.
d) Desenvolver, em paralelo, uma prova em branco e anotar o volume (V4) consumido.
e) Calcular a porcentagem em massa de MgO, mediante a expressão:
M = concentração da solução de EDTA, em mol L-1.
V1 = volume (mL) da solução de EDTA padronizada gasto na titulação do cálcio.
V2 = volume (mL) da solução de EDTA padronizada gasto na titulação da prova em branco do cálcio.
V3 = volume (mL) da solução de EDTA padronizada gasto na titulação do cálcio mais magnésio.
V4 = volume (mL) da solução de EDTA padronizada gasto na titulação da prova em branco do cálcio mais magnésio.
G = peso inicial da amostra, em gramas.
NOTA 211:
i. A relação estequiométrica EDTA-metal nas análises complexométricas é sempre 1:1, seja qual for o número de oxidação do metal.
ii. Os complexos metal-indicador são relativamente estáveis e a viragem pode ser demorada. Sendo assim, as últimas gotas de EDTA devem ser adicionadas lentamente e deve-se cuidar para não ultrapassar o ponto de viragem.
iii. Os indicadores complexométricos são muitas vezes sensíveis à ação do ar e a solução pode tornar-se mais clara durante a titulação. Devem ser adicionadas, então, pequenas quantidades do indicador. Acontece principalmente com o negro de eriocromo T e o calcon.
iv. A viragem deve ser observada horizontalmente, através da solução e o recipiente contendo o analito deve ser colocado em uma posição favorável em relação à luz.
v. As soluções contendo cianeto não podem ser descartadas sem tratamento. Sugestão: oxidação pelo hipoclorito de sódio após alcalinização. Cuidado: nunca adicionar estas soluções a meios ácidos.
¶ 3. ÓXIDO DE CÁLCIO - Método por espectrometria de absorção atômica
O método se baseia na determinação do teor de cálcio a partir da extração em solução de HCl 0,5 ± 0,01 mol L-1 semelhante à descrita na determinação do Poder de Neutralização (PN), por espectrometria de absorção atômica, expressando-se o resultado como óxido de cálcio (CaO). Aplicável de modo geral e mais indicado a produtos com o teor de óxido de cálcio menor ou igual a 7 % em massa (Ca ≤ 5 %). Aplicados a calcários, que geralmente possuem teores de CaO acima desse valor, irá exigir cuidadosa execução, especialmente nas operações de diluição. Pode-se, também, utilizar linhas de ressonância secundárias do cálcio, de modo a minimizar as diluições. Por outro lado, este método é menos susceptível a interferências de outros metais quando comparado com o método do EDTA.
a) Solução de lantânio, com 50 g L-1:tomar 29,33 g de óxido de lantânio, La2O3, p.a., em um béquer de 400 mL e adicionar vagarosamente 250 mL de HCl (1+1), para dissolver o óxido. Transferir para um balão de 500 mL e completar o volume com água.
b) Solução padrão estoque de cálcio, contendo 1000 mg L-1 de Ca: secar carbonato de cálcio (CaCO3, padrão primário) a 285 ± 10 ºC, durante 2 horas, ou seguindo-se a recomendação do fabricante/produtor quanto a secagem do material, e manter em dessecador. Pesar uma massa em gramas igual a [2,4973(100/P)] onde P é a pureza do sal utilizado em porcentagem em massa, transferir para um béquer de 250 mL e dissolver com 20 mL de solução de HCl (1+5). Transferir para balão volumétrico de 1 litro e completar o volume com água. Alternativamente pode ser utilizada solução certificada adquirida pronta para o uso, com rastreabilidade e grau de pureza analítica adequados.
c) Solução padrão intermediária contendo 50 mg L-1:transferir 25 mL da solução de 1000 mg L-1 para um balão volumétrico de 500 mL e completar o volume com HCl (1+23).
d) Soluções padrões de leitura contendo 2,5 - 5- 7,5 e 10 mg L-1 de Ca: transferir para balões de 50 mL: 2,5 - 5 - 7,5 e 10 mL da solução com 50 mg L-1. Adicionar 10 mL da solução de lantânio a todos os balões e completar o volume com água ou HCl (1+23). Preparar um “branco” com água e 10 mL da solução de lantânio também em balão volumétrico de 50 mL.
a) Pesar, com precisão de 0,1 mg, uma massa da amostra de 0,5 g. Deve-se tomar a parte da amostra que foi secada, moída e passada em peneira de 0,30 mm. Esta massa da amostra será identificada nos cálculos como “G”.
b) Transferir para erlenmeyer de 125 mL, adicionar exatamente 25 mL da solução de HCl 0,5 mol L-1 padronizada, cobrir com vidro de relógio e ferver suavemente por 5 minutos. No caso de calcário calcinado ou cal hidratada, aumentar a quantidade de HCl 0,5 mol L-1 para 50 mL, mantendo-se a massa de 0,5 g de amostra.
c) Esfriar, transferir filtrando em papel de filtro de porosidade média para balão volumétrico de 250 mL e lavar o retido com porções de água, deixando cada porção percolar completamente pelo papel de filtro antes de acrescentar a próxima, até um volume de aproximadamente 200 mL. Acrescentar 10 mL de HCl concentrado, completar o volume com água e homogeneizar bem.
NOTA 212: O extrato da amostra com uma relação massa da amostra x volume de 0,5 g para 250 mL propiciará menores diluições no procedimento por espectrometria de absorção atômica.
a) Tomar uma alíquota (A) da solução que contenha até 350 microgramas de óxido de cálcio e transferir para balão de 25 mL. Se necessário, fazer uma diluição intermediária e considerar o fator de diluição (D) no cálculo. Por exemplo, para uma diluição intermediária de 5 mL: 100 mL, com água, o fator D será igual a 20.
NOTA 213: Deve-se tomar uma alíquota de modo a situar a concentração esperada da solução final de leitura, na faixa intermediária da curva de calibração.
b) Adicionar 5,0 mL da solução de óxido de lantânio e completar o volume com água ou HCl (1+23).
c) Colocar o aparelho nas condições exigidas para a determinação do cálcio (lâmpada de Ca, comprimento de onda, fenda e chama adequadas, conforme manual do equipamento).
d) Calibrar o aparelho com o branco e os padrões. Aspirar água entre as leituras e aguardar a estabilização de cada leitura antes de registrar o resultado.
e) Proceder à leitura das soluções das amostras e da prova em branco, verificando a calibração a cada grupo de 8 a 12 leituras e determinar sua concentração em mg L-1através da curva de calibração, equação de regressão ou informação direta do equipamento.
f) Calcular a porcentagem em massa de Ca pela expressão:
C= concentração do elemento obtida na solução de leitura, em mg L-1;
Vbe= volume do balão da extração, em mililitros;
Vbl= volume do balão de leitura, mililitros;
D= fator da diluição intermediária, se houver;
G= massa da amostra, em gramas;
A = alíquota do balão de leitura, mililitros;
10000 = fator de conversão de mg kg-1 para percentagem.
Para expressar o resultado da análise na forma de óxido de cálcio (CaO), fazer a conversão do resultado obtido na análise por meio do seguinte fator:
NOTA 214: Alternativamente as leituras previstas para o equipamento de absorção atômica poderão ser feitas utilizando-se de um espectrômetro de emissão ótica com plasma indutivamente acoplado (ICP-OES), ou espectrômetro de emissão atômica com plasma induzido por micro-ondas (MP-AES), respeitadas as condições de operação do equipamento e a adequação das concentrações das soluções de leitura (padrões e amostras) aos limites de detecção e quantificação específicos para o cálcio.
¶ 4. ÓXIDO DE MAGNÉSIO – Método por espectrometria de absorção atômica
O método se baseia na determinação do teor de magnésio a partir do extrato-amostra da determinação do poder de neutralização (PN), por espectrometria de absorção atômica. Os resultados são apresentados como porcentagem em massa de seu óxido (MgO). Aplicável de modo geral e mais indicado a produtos com teores de MgO da ordem de 8% em massa ou abaixo o que corresponde a Mg ≤ 4,8 %.
Nos produtos em que os teores de óxido de magnésio forem mais elevados (calcários dolomíticos), serão necessárias cuidadosas diluições em duas etapas. Por outro lado, este método é menos susceptível a interferências de outros metais.
a) Solução de lantânio, com 50 g L-1: tomar 29,33 g de óxido de lantânio, La2O3, p.a., em um béquer de 400 mL e adicionar vagarosamente 250 mL de HCl (1+1), para dissolver o óxido. Transferir para um balão de 500 mL e completar o volume com água.
b) Solução padrão estoque de magnésio com 500 mg L-1:Preparar a partir de uma solução padrão certificada de magnésio, adquirida pronta ou a partir de padrões primários, como magnésio metálico, p.a.
c) Solução padrão intermediária de magnésio com 25 mg L-1:tomar 10 mL da solução com 500 mg L-1e diluir em balão volumétrico de 200 mL com ácido clorídrico (1+23). Homogeneizar.
d) Soluções padrões de leitura: transferir 0,5 – 1,0 – 1,5 e 2,0 mL da solução com 25 mg L-1para balão volumétrico de 25 mL. Adicionar 5 mL da solução de lantânio a todos os balões e completar o volume com água ou HCl (1+23). Estas soluções contêm 0,5 - 1,0 – 1,5 e 2,0 mg L-1. Preparar um “branco” com água ou HCl (1+23) e 5 mL da solução de lantânio também em balão volumétrico de 25 mL.
Utilizar o extrato-amostra preparado para a determinação do poder de neutralização (PN), conforme descrito em 4.C.1.4.
a) Tomar uma alíquota (A) do extrato que contenha no máximo 80 microgramas de óxido de magnésio e transferir para balão de 25 mL.
NOTA 215:
i. Deve-se tomar uma alíquota de modo a situar a concentração esperada da solução final de leitura na faixa intermediária da curva de calibração.
ii. Pode ser necessário fazer uma diluição intermediária, com HCl (1+23), considerando-se, no cálculo final o fator de diluição “D”. Por exemplo, para uma diluição de 5:100, o fator de diluição D = 20.
b) Adicionar 5 mL de óxido de lantânio a 50 g L-1, completar o volume com água ou HCl (1+23) e homogeneizar.
c) Colocar o aparelho nas condições exigidas para a determinação do magnésio (lâmpada de Mg, comprimento de onda, fenda e chama adequadas, conforme manual do equipamento).
d) Calibrar o aparelho com o branco e os padrões. Aspirar água entre as leituras e aguardar a estabilização de cada leitura antes de registrar o resultado.
e) Proceder à leitura das soluções das amostras e da prova em branco, verificando a calibração a cada grupo de 8 a 12 leituras e determinar sua concentração em mg L-1 através da curva de calibração, equação de regressão ou informação direta do equipamento.
f) Calcular a porcentagem em massa de Mg pela expressão:
C = concentração de Mg na solução final de leitura, em mg L-1.
Vb = volume do balão utilizado na etapa de extração, em mililitros.
D = fator de diluição intermediária do extrato inicial, se tiver ocorrido.
A = volume da alíquota tomada para a solução de leitura, em mililitros.
G = massa inicial da amostra, em g.
Para expressar o resultado da análise na forma de óxido de magnésio (MgO), fazer a conversão do resultado obtido na análise por meio do seguinte fator:
NOTA 216: Alternativamente as leituras previstas para o equipamento de absorção atômica poderão ser feitas utilizando-se de um espectrômetro de emissão ótica com plasma indutivamente acoplado (ICP-OES), ou espectrômetro de emissão atômica com plasma induzido por micro-ondas (MP-AES), respeitadas as condições de operação do equipamento e a adequação das concentrações das soluções de leitura (padrões e amostras) aos limites de detecção e quantificação específicos para o magnésio.
Para corretivos de acidez que tenham uma composição com mais impurezas, presença de outros metais e mesmo fosfato podem ser empregados os métodos descritos a seguir, que são de execução mais laboriosa: o método permanganométrico para a determinação do óxido de cálcio e o método gravimétrico do pirofosfato para a determinação do óxido de magnésio.
¶ 5.1. ÓXIDO DE CÁLCIO - Método volumétrico do permanganato de potássio
O método baseia-se na precipitação do cálcio como oxalato de cálcio, solubilização deste com ácido sulfúrico, formando-se o ácido oxálico, que será titulado com uma solução padronizada de permanganato de potássio.
c) Solução de bromofenol azul a 0,2 % (m/v): tomar 0,10 g de bromofenol azul em uma cápsula de porcelana, adicionar 3 mL de uma solução aquosa de NaOH a 0,2 % (m/v), aos poucos (porções de 0,2-0,3 mL), homogeneizando até dissolver o material sólido. Transferir para um balão volumétrico de 50 mL, completar o volume com água e homogeneizar. Utilizar reagentes p.a.
d) Solução de hidróxido de amônio (1 + 4),com água.
e) Solução de ácido clorídrico (1 + 4), com água.
f) Solução saturada de oxalato de amônio: suspender 80 g de (NH4)2C2O4.H2O em 50 mL de água, transferir para balão volumétrico de 1 litro, completar o volume e homogeneizar. Deixar em repouso por 12 – 18 horas. Utilizar reagente p.a.
g) Soluções de ácido sulfúrico – H2SO4 – (1 + 9) e (1 + 19), com água.
h) Oxalato de sódio – Na2C2O4, padrão primário secado a 105 ± 5ºC por 1 hora, ou seguindo-se a recomendação do fabricante/produtor quanto a secagem do material, e conservado em dessecador.
i) Solução de permanganato de potássio, KMnO4 padronizada, com 0,02 mol L-1. Preparo: Dissolver 3,2 g de KMnO4, p.a., em um litro de água destilada, ferver por uma hora, cobrir com vidro de relógio e deixar repousar durante 12 a 18 horas. Filtrar, com sucção, através de funil com placa filtrante de vidro sinterizado de porosidade média (16 a 40 µm) ou fina (10 a 16 µm), e transferir o filtrado para um recipiente de vidro escuro.
Padronizaçãosolução de permanganato de potássio 0,02 mol L-1 :
i) Pesar 0,2 g de oxalato de sódio com precisão de 0,1 mg e transferir para erlenmeyer de 500 mL; acrescentar 250 mL da solução de H2SO4 (1 + 19) previamente fervida por 15 minutos e esfriada em temperatura ambiente.
ii) Transferir a solução de KMnO4 para uma bureta; adicionar 25 mL dessa solução para dentro do erlenmeyer durante 60-90 segundos, com agitação contínua. Deixar em repouso até a cor desaparecer (caso não desapareça, repetir adicionando menor volume de KMnO4).
iii) Aquecer a solução do erlenmeyer a 50-60 ºC e prosseguir a titulação, até uma leve cor rósea persistir por 30 segundos, adicionando o permanganato, no final, gota a gota, esperando cada gota perder completamente a cor antes da adição da próxima.
iv) Calcular a concentração (M) da solução de permanganato, em mol L-1, pela expressão:
m= massa de oxalato de sódio, em gramas.
V= volume da solução de KMnO4 gasto na titulação, em mililitros.
Repetir por mais duas vezes e fazer a média dos valores de concentração obtidos.
Utilizar o extrato-amostra preparado para a determinação do poder de neutralização (PN), conforme descrito em 4.C.1.4.
a) Transferir uma alíquota (A) do extrato que contenha de 15 a 80 mg de CaO provável para um béquer de 300- 400 mL, adicionar 70-80 mL de água destilada e homogeneizar.
b) Adicionar 4 gotas da solução de bromofenol azul e solução de amônia (1 + 4), aos poucos, até o indicador passar da cor amarela a verde (pH 3,5 a 4,0). Em seguida, adicionar mais 40-50 mL de água.
NOTA 217:
i. Pode ser usado o indicador vermelho de metila em solução alcoólica a 0,2 % m/v (0,2 g do indicador em 100 mL de álcool etílico p.a.) e a mudança de cor deverá ser de vermelho para rosa (pH 3,5-4,0).
ii. O pH deve ser mantido na faixa de 3,5-4,0. Se a coloração mudar de verde para azul ou voltar a amarelo novamente, corrigir com HCl (1+4) ou NH4OH (1+4), respectivamente.
c) Aquecer até quase atingir a ebulição e adicionar, aos poucos, 30 mL da solução saturada de oxalato de amônio a 85-90 ºC, agitando continuamente. Verifica-se a precipitação do oxalato de cálcio.
NOTA218: Nessa operação, manter o pH da solução indicada pela cor verde do indicador (ou cor rósea, se o indicador for vermelho de metila) através do emprego da solução de NH4OH (1+4) ou de HCl (1 + 4).
d) Deixar em banho-maria durante 1 hora e esfriar, mantendo sempre o pH indicado.
e) Filtrar através do papel de filtro de porosidade média ou de cadinho com fundo de vidro sinterizado de porosidade média (16 a 40 µm), recebendo o filtrado em um erlenmeyer de 300 mL ou em um frasco de filtração a vácuo, de 300 mL.
f) Lavar o precipitado com 10 porções de água quente (70-80ºC), de 10 mL cada uma.
g) Retirar o recipiente (erlenmeyer ou o frasco de filtração a vácuo) que recebeu o filtrado, conservando o seu conteúdo para uma possível determinação gravimétrica do magnésio, e substituir por outro similar.
h) Dissolver o oxalato de cálcio com 10 porções, de 10 mL cada, da solução de H2SO4 (1+ 9) a 70-80ºC.
i) Titular a 70-80ºC com a solução padronizada de permanganato de potássio 0,02 mol L-1.
j) Desenvolver, em paralelo, uma prova em branco.
k) Calcular o porcentual em massa de CaO pela expressão:
V1= volume da solução de permanganato gasto na titulação da amostra, em mililitros.
V2= volume da solução de permanganato gasto na titulação da prova em branco, em mililitros.
M= concentração da solução de KMnO4, em mol L-1.
G= massa da amostra contida na alíquota (A) tomada no item “a”, em gramas.
NOTA219: 1 mL de KMnO4 0,02 mol L-1 equivale a 2 mg de Ca e 2,8 mg de CaO.
¶ 5.2. ÓXIDO DE MAGNÉSIO - Método gravimétrico do pirofosfato
O método baseia-se na precipitação do magnésio como fosfato duplo de amônio e magnésio hexahidratado – MgNH4PO4.6H2O – seguindo-se a calcinação do precipitado a pirofosfato de magnésio – Mg2P2O7 – forma sob a qual será pesado para a determinação gravimétrica. Aplica-se aos corretivos de acidez com teor de MgO ≥ 8%.
b) Solução de bromofenol azul a 0,2 % (m/v): tomar 0,10 g de bromofenol azul em uma cápsula de porcelana, adicionar 3 mL de uma solução aquosa de NaOH a 2% (m/v), aos poucos (porções de 0,2-0,3 mL), homogeneizando até dissolver o material sólido. Transferir para um balão volumétrico de 50 mL, completar o volume com água e homogeneizar. Utilizar reagentes p.a.
c) Hidróxido de amônio (NH4OH), p.a.
d) Soluções de NH4OH e água nas relações (1 + 1), (1 + 4) e (1+ 9).
e) Solução saturada de oxalato de amônio: suspender 80 g de (NH4)2C2O4.H2O em 50 mL de água, transferir para balão volumétrico de 1 litro, completar o volume e homogeneizar. Deixar em repouso por 12 – 18 horas.
f) Solução de ortofosfato diamônio a 20 % (m/v): dissolver 20,0 g de (NH4)2HPO4 em 100 mL de água.
g) Solução de ácido cítrico (C6H8O7.H2O), a 10% (m/v): dissolver 10 g de ácido cítrico mono-hidratado em 70-80 mL de água e completar o volume a 100 mL.
h) Solução de bromotimol azul a 0,2 % (m/v): tomar 0,10 g de bromotimol azul em uma cápsula de porcelana, adicionar 3,2 mL de uma solução aquosa de NaOH a 2% (m/v) aos poucos (proporções de 0,20-0,30 mL), homogeneizando até dissolver o material sólido. Transferir para um balão volumétrico de 50 mL, completar o volume com água destilada e homogeneizar.
Utilizar o extrato-amostra preparado para a determinação do poder de neutralização (PN), conforme descrito em 4.C.1.4.
a) Transferir uma alíquota (A) do extrato que contenha de 30 a 160 mg de MgO provável para um béquer de 300- 400 mL, adicionar 70-80 mL de água destilada e homogeneizar.
b) Adicionar 4 gotas de solução bromofenol azul e solução de amônia (1 + 4), aos poucos, até o indicador passar da cor amarela a verde (pH 3,5 a 4,0). Em seguida, adicionar mais 40-50 mL de água destilada.
NOTA 220:
i. Pode ser usado o indicador vermelho de metila em solução alcoólica a 0,2 % m/v (0,2 g do indicador em 100 mL de álcool etílico p.a.) e a mudança de cor deverá ser de vermelho para rosa (pH 3,5-4,0).
ii. O pH deve ser mantido na faixa de 3.5-4,0. Se a coloração mudar de verde para azul ou voltar a amarelo novamente, corrigir com HCl (1+4) ou NH4OH (1+4), respectivamente.
c)Aquecer até quase atingir a ebulição e adicionar, aos poucos, 30 mL de solução saturada de oxalato de amônio a 85-90 ºC, agitando continuamente. Verifica-se a precipitação do oxalato de cálcio.
NOTA 221: Nessa operação, manter o pH da solução indicada pela cor verde do indicador (ou cor rósea, se o indicador for vermelho de metila) através do emprego da solução de NH4OH (1 + 4) ou de HCl (1 + 4).
d) Deixar em banho-maria durante 1 hora e esfriar, mantendo sempre o pH indicado.
e) Filtrar através do papel de filtro de porosidade média ou de cadinho com fundo de vidro sinterizado de porosidade média (16 a 40 µm), para um erlenmeyer de 300 mL ou um frasco de filtração a vácuo, de 300 mL.
f) Lavar o precipitado com 10 porções de água quente (70-80 ºC), de 10 mL cada uma.
NOTA 222: Este procedimento até este ponto é o mesmo da determinação do Óxido de Cálcio com permanganato – Método 4.C.5.1 anterior – podendo ser executado apenas uma vez para as duas determinações.
g) Transferir para um béquer de 400 mL o filtrado obtido da separação do oxalato de cálcio.
h) Adicionar ao filtrado 10 mL da solução de ácido cítrico a 10%, 4 gotas da solução de bromotimol azul, solução de NH4OH (1 + 1) até a viragem do indicador (a solução deverá ficar azul) e 10 mL da solução do ortofosfato diamônico a 20 % m/v.
i) Agitar vigorosamente a solução com o auxílio de um bastonete de vidro sem encostar ou atritar as paredes do copo até a formação de precipitado.
j) Adicionar 15 mL de NH4OH concentrado, deixar em repouso por 2 horas, agitando 2 a 3 vezes na primeira hora (quando a quantidade de precipitado for muito pequena ou quando não se percebe a sua formação, deixar em repouso durante a noite).
k) Filtrar através de papel de filtro de filtração lenta, adaptado a um funil de haste longa, para um erlenmeyer de 500 mL ou copo de 600 mL.
l) Lavar o copo em que foi feita a precipitação, o papel de filtro e o precipitado com 10 porções, de 10 mL cada uma, de solução de NH4OH (1 + 9).
m) Transferir o papel de filtro contendo o precipitado para um cadinho de porcelana, previamente tarado, colocar o cadinho na entrada da mufla a 850-900ºC e deixar até queimar o papel. Transferir o cadinho para o centro do forno e deixar a 900ºC durante uma hora.
n) Retirar o cadinho da mufla, colocá-lo em dessecador, deixar esfriar e pesar.
o) Calcular o percentual em massa de MgO pela expressão:
onde:
P= massa do precipitado (Mg2P2O7), em gramas.
G= massa da amostra, contido na alíquota (A) tomada no item “a”, em gramas.
O método consiste na extração ácida dos metais contidos na amostra e sua determinação em espectrômetro de absorção atômica (EAA) ou, alternativamente, em espectrômetro de emissão óptica com plasma indutivamente acoplado (ICP-OES) ou espectrômetro de emissão atômica com plasma induzido por micro-ondas (MP-AES). Aplicável aos corretivos de acidez.
b) Solução aquosa de HCl (1+23), aproximadamente 0,5 mol L-1.
c) Soluções padrões estoque com 1000mg L-1 dos metais Cd e Pb: podem ser utilizadas soluções certificadas adquiridas prontas ou serem preparadas a partir de padrões primários contendo os metais referidos.
d) Soluções de concentração intermediária (100 mg L-1) dos metais, preparadas por diluição da solução-estoque com solução de HCl (1+23).
e) Soluções padrões de leitura, com concentrações de acordo com a faixa de leitura, para cada um dos elementos.
¶ 6.4.1. Extração de corretivos de modo geral, exceto os ricos em silicatos
a) Pesar de 1 a 2 g da amostra (massa “G”) com precisão de 0,1 mg e transferir para um béquer de 150 mL, erlenmeyer de 125 mL ou tubo de digestão apropriado. Deve-se tomar a parte da amostra que foi secada, moída e passada em peneira de 0,30 mm.
b) Acrescentar à amostra 5-10 mL de água, homogeneizar e adicionar, com cuidado, principalmente no início (devido à efervescência), 10 mL de HCl e 3 mL de HNO3 concentrados para cada grama de amostra tomada. Preparar simultaneamente uma prova em branco dessa extração.
c) Cobrir com vidro de relógio, levar ao banho-maria, placa, chapa ou bloco de aquecimento com temperatura controlada e ferver até reduzir o volume a 2-3 mL (estado xaroposo). Esfriar, adicionar 20 mL de água e 5 mL de HCl concentrado. Ferver por 10 minutos e deixar esfriar ligeiramente para permitir o manuseio. Filtrar com papel de filtro de porosidade média (ou fina, se necessário) para balão volumétrico de 100 mL ou de um volume Vb mais adequado, de acordo com a concentração do contaminante na amostra, de modo a minimizar as operações de diluição.
d) Lavar o retido com água quente (80-90 ºC), deixar esfriar e completar o volume. Homogeneizar, obtendo-se o extrato-amostra.
e) Fazer as diluições necessárias para leitura, utilizando soluções aquosas de ácido clorídrico (1+23) para leitura em espectrômetro de absorção atômica.
¶ 6.4.2. Extração de corretivos ricos em silicatos
¶6.4.2.1. Extração com ácido perclórico e ácido fluorídrico
Esta operação não pode ser realizada em recipiente de vidro ou porcelana, devendo-se usar cadinhos de platina ou de teflon. O manuseio dos ácidos fluorídrico e perclórico deve ser feito com bastante cuidado (luvas, óculos e demais EPI’s).
a) Pesar uma massa (G) de 0,5 a 1 g da amostra, com precisão de 0,1 mg, transferir para cadinho de platina ou de teflon e acrescentar 5 mL de HClO4 e 5 mL de HF concentrados. Conduzir, em paralelo, uma prova em branco.
b) Colocar o cadinho em uma cápsula de porcelana de fundo chato e o conjunto sobre uma chapa aquecedora.
c) Aquecer até o desprendimento de densos vapores brancos de HClO4. (Cuidado para não deixar secar).
d) Retirar da chapa, deixar esfriar e transferir quantitativamente para um béquer de 150 mL, fazendo um volume de aproximadamente 50 mL, com água. Aquecer, levando a ebulição moderada por 10 minutos. Deixar esfriar.
e) Transferir quantitativamente para um balão volumétrico de 100 mL (Vb). Para produtos concentrados este volume final poderá ser aumentado, de modo a permitir menores diluições para a leitura no espectrômetro de absorção atômica. Completar o volume com água e homogeneizar.
f) Filtrar em papel de filtro de porosidade média ou fina, se necessário, recebendo o filtrado em um recipiente seco.
¶6.4.2.2. Extração em sistema aberto (método AOAC 965.09 item c-alternativo)
a) Pesar uma massa (G) de 0,5 g da amostra, com precisão de 0,1 mg, e transferir para cápsula de teflon ou cadinho de platina. Adicionar 10 mL de HCl, 5 mL de HF e 10 mL de metanol, nessa ordem.
b) Aquecer a amostra até secura. Em seguida, adicionar 5 mL de HCl e novamente levar a amostra à secura. Repetir a adição de 5 mL de HCl e novamente levar a amostra à secura. Depois, adicionar 20 mL da solução de HCl (1+5)e ferver brandamente a amostra por 5 minutos.
c) Deixar esfriar e transferir quantitativamente para balão de volumétrico de 100 mL (Vb). Completar o volume com água e homogeneizar.
d) Filtrar em papel de filtro de porosidade média ou fina, se necessário, recebendo o filtrado em um recipiente seco.
¶6.4.2.3. Extração assistida por forno de micro-ondas (método EPA 3052)
a) Pesar uma massa (G) de 0,25 g de amostra, com precisão de 0,1 mg, e transferir para tubo de Teflon do forno de micro-ondas.
b) Adicionar 9 mL de HNO3 e 4 mL de HF concentrados e deixar reagir por 15 minutos com o tubo digestor aberto dentro da capela. Preparar simultaneamente uma prova em branco dessa extração.
c) Proceder à digestão em aparelho de micro-ondas conforme manual do equipamento, utilizando a seguinte programação de aquecimento:
Sequência
Temperatura (ºC)
Tempo (min)
Potência (%)
1
180 ± 5
5,5
90
2
180 ± 5
9,5
90
d) Após o resfriamento das amostras, retirar os suportes do rotor, e vagarosamente abrir os tubos de Teflon, para evitar perda de amostra na liberação dos gases.
e) Após esfriarem, transferir as amostras para cadinho de teflon e aquecer até secura em chapa aquecedora, para eliminação do HF residual. Na sequência, adicionar 20 mL de solução HCl (1+5) e levar a amostra à ebulição branda por 5 minutos.
f) Transferir quantitativamente cada amostra para balão volumétrico de 50 mL (Vb) ou outro volume apropriado.
g) Filtrar em papel de filtro de porosidade média ou fina, se necessário.
a) Preparar os padrões de leitura, por diluições da solução intermediária, seguindo as recomendações de faixa de concentração que garanta a linearidade da curva, comprimento de onda e tipo de chama indicados no manual do equipamento.
NOTA 223: Caso o laboratório tenha a disponibilidade de uso de micropipetas, fica facultativo o preparo dos padrões da curva de calibração a partir de soluções intermediárias, podendo ser preparados diretamente das soluções padrões estoque.
b) Colocar o equipamento nas condições operacionais adequadas para a obtenção das leituras.
Sugestões de condições operacionais
Para cádmio:
Chama ar x acetileno oxidante. A absorbância é fortemente dependente do ajuste correto da corrente da lâmpada e estequiometria da chama.
Comprimento de onda 228,8 nm.
Para chumbo:
Chama ar x acetileno oxidante.
Utilizando o comprimento de onda de 217 nm: faixa de 0 a 15 mg L-1.
Trabalhando no comprimento de onda de 283,3 nm: faixa de 0 a 30 mg L-1.
c) Feitas as leituras dos padrões, montar a curva de calibração e calcular a equação de regressão.
a) Conduzir a leitura da prova em branco (matriz de abertura da amostra) para subtrair do valor de leitura das amostras.
b) Tomar uma alíquota (A) do extrato-amostra e transferir para balão volumétrico de volume Vc, de modo que a concentração final da solução de leitura esteja no intervalo de concentração dos padrões, de preferência na faixa média da curva de calibração para cada elemento.
c) Proceder às leituras e registrá-las. Converter as leituras encontradas para as concentrações correspondentes através da equação de regressão linear ou obtê-las por informação direta do equipamento utilizado. A partir das concentrações, calcular o teor nas amostras, reportando-se à massa (G) tomada inicialmente.
d) Fórmula geral de cálculo:
E: teor do elemento (Cd ou Pb) na amostra, em mg kg-1.
C: concentração do elemento na solução de leitura, em mg L-1.
Cb: concentração da prova em branco, em mg L-1.Vc: volume do balão volumétrico da solução de leitura.
Vb: volume do balão volumétrico utilizado na preparação do extrato-amostra.
G: massa inicial da amostra, em gramas.
A: alíquota tomada para a solução de leitura, em mililitros.
NOTA 224: A leitura poderá, também, ser feita diretamente no extrato-amostra:
Se, ao contrário, for necessária uma diluição intermediária, multiplicar pelo fator de diluição.
NOTA 225: Alternativamente as leituras previstas para o equipamento de absorção atômica poderão ser feitas utilizando-se de um espectrômetro de emissão ótica com plasma indutivamente acoplado (ICP-OES), ou espectrômetro de emissão atômica com plasma induzido por micro-ondas (MP-AES), respeitadas as condições de operação do equipamento e a adequação das concentrações das soluções de leitura (padrões e amostras) aos limites de detecção e quantificação específicos para os elementos cádmio e chumbo.
¶ D – CÁLCULO DO PODER RELATIVO DE NEUTRALIZAÇÃO TOTAL (PRNT)
5.1.1 LEI Nº 14.515, DE 29 DE DEZEMBRO DE 2022 - Dispõe sobre os programas de autocontrole dos agentes privados regulados pela defesa agropecuária e sobre a organização e os procedimentos aplicados pela defesa agropecuária aos agentes das cadeias produtivas do setor agropecuário; institui o Programa de Incentivo à Conformidade em Defesa Agropecuária, a Comissão Especial de Recursos de Defesa Agropecuária e o Programa de Vigilância em Defesa Agropecuária para Fronteiras Internacionais (Vigifronteiras); altera as Leis nºs 13.996, de 5 de maio de 2020, 9.972, de 25 de maio de 2000, e 8.171, de 17 de janeiro de 1991; e revoga dispositivos dos Decretos-Leis nºs 467, de 13 de fevereiro de 1969, e 917, de 7 de outubro de 1969, e das Leis nºs 6.198, de 26 de dezembro de 1974, 6.446, de 5 de outubro de 1977, 6.894, de 16 de dezembro de 1980, 7.678, de 8 de novembro de 1988, 7.889, de 23 de novembro de 1989, 8.918, de 14 de julho de 1994, 9.972, de 25 de maio de 2000, 10.711, de 5 de agosto de 2003, e 10.831, de 23 de dezembro de 2003.
5.1.3 LEI Nº 6.894, DE 16 DE DEZEMBRO DE 1980 - Dispõe sobre a inspeção e a fiscalização da produção e do comércio de fertilizantes, corretivos, inoculantes, estimulantes ou biofertilizantes, remineralizadores e substratos para plantas, destinados à agricultura, e dá outras providências.
ALCARDE, J. C. Metodologia de análise de fertilizantes e corretivos. Piracicaba: Instituto do Açúcar e do Álcool, 1979. 274 p.
ALCARDE, J. C. Métodos simplificados de análise de fertilizantes (N, P, K) minerais. Brasília: Ministério da Agricultura, 1982. 49 p.
ALCARDE, J. C. et al.Avaliação da higroscopicidade de fertilizantes e corretivos. Scientia Agrícola, Piracicaba, v. 49, n. 1, p. 137-144, 1992.
ALCARDE, J. C.; RODELLA, A. A. Caracterização de fertilizantes simples contendo zinco. Scientia Agrícola, Piracicaba, v. 50, n. 1, p. 121-126, 1993.
ALCARDE, J. C.; RODELLA, A. A. O equivalente em carbonato de cálcio dos corretivos da acidez dos solos. Scientia Agrícola, Piracicaba, v. 53, n. 2/3, p. 204-210, 1996.
ALCARDE, J. C.; RODELLA, A. A. Avaliação química de corretivos de acidez para fins agrícolas: uma nova proposição. Scientia Agrícola, Piracicaba, v. 53, n. 2/3, p. 211-216, 1996.
ALCARDE, J. C.; VALE, F. Avaliação química de fertilizantes com micronutrientes comercializados no Brasil, In: Congresso Latino Americano de la Ciencia del Suelo, 14 1999, Pucon, Chile, CLACS99, ANAIS. Temuco: Universidade de La Frontera, 1999, 1 CD-ROM.
ALCARDE, J. C.; VALE, F. Solubilidade de micronutrientes contidos em misturas de fertilizantes, em extratores químicos. Revista Brasileira de Ciência do Solo, Campinas, 27: 363-372, 2003.
ALCARDE, J. C. Manual de análise de fertilizantes, Piracicaba: Fundação de Estudos Agrários “Luiz de Queiróz”, 2009, 259p.
ALCARDE, J. C. Metodologia oficial de análise de corretivos de acidez, 2ª ed., 2009 – Boletim editado pela Associação Brasileira dos Produtores de Calcário (ABRACAL) e Sindicato das Indústrias de Calcário e Derivados para Uso Agrícola no Estado de São Paulo (SINDICAL).
ABREU, M. F.; ANDRADE, J. C.; FALCÃO, A. A. Protocolos de análises químicas. In: ANDRADE, J. C.; ABREU, M. F. Análise química de resíduos sólidos para monitoramento e estudos agroambientais. Campinas: Instituto Agronômico, 2006. cap. 9, p. 121-158.
ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS, Fórum Nacional de Normatização. Águas – determinação da demanda química de oxigênio (DQO): NBR 10357, 1988, 11p.
ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS, Fórum Nacional de Normatização. Fertilizantes orgânicos - determinação do carbono orgânico – Método de Walkey-Black: MB-3806, 1989, 2p.
ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS, NBR 15822:2010, primeira edição 12.04.2010, válida a partir de 12.05.2010: Fertilizantes fluidos, determinação da densidade, método do picnômetro, 4p.
BATAGLIA, O. C.; VAN RAIJ, B. Eficiência de extratores de micronutrientes na análise de solo. Revista Brasileira de Ciência do Solo, Campinas, v.13, p.205-212, 1989.
BENITES, V. M.; MADARI, B.; MACHADO, P. L. O. A. Extração e fracionamento quantitativo de substâncias húmicas do solo: um procedimento simplificado de baixo custo. Rio de Janeiro, 2003. 7p. (Embrapa Solos- Comunicado Técnico, 16).
BENITES, V. M. et al.Comparação de métodos de determinação de carbono por via úmida em solos tropicais. Rio de Janeiro, 2004. 5p. (Embrapa Solos- Circular Técnica, 27).
BISUTTI, I.; HILKE, I.; RAESSLER, M. Determination of total organic carbon – an overview of current methods. Trends in Analytical Chemistry, Amsterdam, v.23, n. 10-11, p. 716-726, 2004.
CAMARGO, F. A. O.; SANTOS, G. A.; GUERRA, J. G. M. Macromoléculas e substâncias húmicas. In: SANTOS, G. A.; CAMARGO, F. A. O. Fundamentos da matéria orgânica do solo: ecossistemas tropicais e subtropicais. Porto Alegre: Genesis, 1999. cap. 3.
CIAVATTA, C.; VITTORI ANTISARI, L.; SEQUI, P. Determination of organic carbon in soils and fertilizers. Communications In Soil Science and Plant Analysis, v. 20, n.7-8 p. 759 - 773, 1989.
DIAS, L. E. et al.Comparação de diferentes métodos de determinação de carbono orgânico em amostras de solos. Revista Brasileira de Ciência do Solo, Campinas, v.15, p.157-162, 1991.
JEFFERY, G. H. et al.Análise química quantitativa. 5. ed. Rio de Janeiro: LTC, 1992. 712 p.
KANE, P. F. Fertilizers. In: HORWITZ, W. (Ed.). Official methods of analysis of AOAC International. 17. ed. Gaithersburg: AOAC International, 2000. v. 1.
KORNDÖRFER, G. H.; PEREIRA, H. S.; NOLLA, A. Análise de silício: solo, planta e fertilizante. Uberlândia: GPSI, ICIAG/UFU, 2004. (GPSI- Boletim Técnico, 2).
MATSUO, H.; MIYASAKI, Y.; TAKEMURA.; MATSUOKA, S.; SAKASHITA, H.; YOSHIMURA, K.B. NMR study on the interation of boric acid with Azomethine H. Polyhedron, Vol.23, 995-961, 2004.
MENDONÇA, E. S.; MATOS, E. S. Matéria orgânica do solo: métodos de análises. Viçosa: UFV, 2005. 107 p.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Decreto N° 4.954 de 14 de janeiro de 2004, alterado pelo Decreto nº 8.384, de 29 de dezembro de 2014. Aprova o Regulamento da Lei nº 6.894, de 16 de dezembro de 1980, que dispõe sobre a inspeção e fiscalização da produção e do comércio de fertilizantes, corretivos, inoculantes, biofertilizantes, remineralizadores e substratos para plantas destinados à agricultura, e dá outras providências. Diário Oficial da União, 15 de janeiro de 2004. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Ministério da Agricultura. Análises de corretivos, fertilizantes e inoculantes - Métodos oficiais. Brasília, Laboratório Nacional de Defesa Agropecuária, 1983, 104p.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Laboratório Nacional de Referência Vegetal. Análise de corretivos, fertilizantes e inoculantes: métodos oficiais. Brasília: LANARV, 1988. 104 p.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa nº 05, de 10 de março de 2016. Dispõe sobre as definições, classificação, especificações e garantias, tolerâncias, registro, embalagem, rotulagem e propaganda dos remineralizadores e substratos para plantas, destinados à agricultura. Diário Oficial da União nº 49, de 14 de março de 2016, Seção 1, p. 10. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa nº 24, de 20 de junho de 2007. Reconhece métodos analíticos constantes do Anexo da IN, para a determinação de metais pesados tóxicos em fertilizantes, corretivos agrícolas, condicionadores de solo e substratos para plantas. Diário Oficial da União de 21 de junho de 2007, Seção 1. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa nº 61, de 8 de julho de 2020. Estabelece as regras sobre definições, exigências, especificações, garantias, tolerâncias, registro, embalagem e rotulagem dos fertilizantes orgânicos e dos biofertilizantes, destinados à agricultura. Diário Oficial da União de 15 de julho de 2020, Seção 1, p. 5. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa nº 27, de 05 de junho de 2006, alterada pela IN SDA nº 07, de 12 de abril de 2016. Dispõe sobre os limites estabelecidos para agentes fitotóxicos, patogênicos ao homem, animais e plantas, metais pesados tóxicos, pragas e ervas daninhas, presentes em fertilizantes, corretivos, inoculantes e biofertilizantes. Diário Oficial da União de 13 de abril de 2016, Seção 1. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa nº 28, de 27 de julho de 2007. Aprova os métodos analíticos oficiais para fertilizantes minerais, orgânicos, organominerais e corretivos, constantes do Anexo da IN. Diário Oficial da União de 31 de julho de 2007, Seção 1. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa nº 35, de 04 de julho de 2006. Dispõe sobre as normas sobre especificações e garantias, tolerâncias, registro, embalagem e rotulagem dos corretivos de acidez, de alcalinidade e de sodicidade e dos condicionadores de solo, destinados à agricultura. Diário Oficial da União de 12 de julho de 2006, Seção 1, p. 32. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa nº 39, de 8 de agosto de 2018. Dispõe as regras sobre definições, exigências, especificações, garantias, registro de produto, autorizações, embalagem, rotulagem, documentos fiscais, propaganda e tolerâncias dos fertilizantes minerais destinados à agricultura. Diário Oficial da União nº 154, de 10 de agosto de 2018, Seção 1, p. 19. Brasília, DF.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa GM nº 53, de 23 de outubro de 2013, alterada pela IN SDA/Mapa nº 06, de 10 de março de 2016. Dispõe sobre as definições, classificação, registro e renovação de registro de estabelecimentos, cadastro e renovação de cadastro de prestadores de serviços, fornecedores de minérios e geradores de materiais secundários, alterações, cancelamentos e outros. Diário Oficial da União nº 49, de 14 de março de 2016, Seção 1, p. 11. Brasília, DF.
Official Methods of Analysis of AOAC INTERNATIONAL (1980), AOAC INTERNATIONAL, Gaithersburg, MD, USA, Official Method 960.04.
Official Methods of Analysis of AOAC INTERNATIONAL (1980), AOAC INTERNATIONAL, Gaithersburg, MD, USA, Official Method 976.01.
OLIVEIRA, E. A. B. Avaliação de método alternativo para extração e racionamento de substâncias húmicas em fertilizantes orgânicos. 2011. 48 f. Dissertação (Mestrado em Agricultura Tropical e Subtropical) – Instituto Agronômico, Campinas, 2011.
OLIVEIRA, S. C. Solubilidade e disponibilidade de micronutrientes e metais pesados tóxicos em fertilizantes comercializados no Brasil. Dissertação (Mestrado) - Escola Superior de Agricultura Luiz de Queiroz, Piracicaba, 2003. 156p.
ORGANISATION, Federal Compost Quality Assurance. Methods Book for the Analysis of Compost. Germany: Bundesgütegemeinschaft Kompost eV, 2003, p. 45.
RODELLA, A. A.; ALCARDE, J. C. Avaliação de materiais orgânicos empregados como fertilizantes. Scientia Agrícola, Piracicaba, v. 51, n.3, p. 556-562, 1994.
RODELLA, A. A. Análise química de fosfito. Texto didático. Dados não publicados. 6 p, s.d.
SANTIAGO, A. R. de O. Avaliação e validação do método espectrofotométrico da azomethina-H para determinação do boro em fertilizantes com conteúdo orgânico. 2013. 77 f. Trabalho de Conclusão de Curso (Química) - Instituto Federal de Educação, Ciência e Tecnologia, Goiás, 2013.
SLEUTEL S.; de NEVE S.; SINGIER B; HOFMAN, G. Quantification of Organic Carbon in Soils: A Comparison of Methodologies and Assessment of the Carbon Content of Organic Matter. Communications in Soil Science and Plant Analysis. v. 38, p. 2647–2657, 2007.
SNELL, F. D.; ETTRE, L. S. (Ed.). Encyclopedia of industrial chemical analysis. New York: Intercience, 1973. v. 18. 545 p.
THE NATIONAL INSTITUTE OF AGRICULTURAL SCIENCES (Japan). Official methods of analysis of fertilizers. Nishigahara: Ministry of Agriculture and Forestry, 1977. 116 p.
TREVIZAM, A. R. Solubilidade de micronutrientes e elementos contaminantes em fertilizantes. Piracicaba, 2005. Dissertação (Mestrado) – Centro de Energia Nuclear na Agricultura, Universidade de São Paulo, Piracicaba, 2005. 132p.
UNIÃO EUROPÉIA. Regulamento (CE) nº 2003/2003 do Parlamento Europeu e do Conselho de 13 de outubro de 2003 relativo aos adubos. Jornal Oficial da União Européia, Luxemburgo, L 304 de 21 nov. 2003. 194 p.
VAN RAIJ, B. et al.Análise química para avaliação da fertilidade de solos tropicais. Campinas: Instituto Agronômico, 2001. 285 p.
VALE, F. & ALCARDE, J. C. Extratores para avaliar a disponibilidade do zinco em fertilizantes. Revista Brasileira de Ciência do Solo, Campinas, v. 26, p. 655-662, 2002.
VALE, F.; ALCARDE, J. C. Solubilidade e disponibilidade dos micronutrientes em fertilizantes. Revista Brasileira de Ciência do Solo, Campinas, v. 23, p. 441-451, 999.
Cavins, T.J.; Whipker, B.E.; Fonteno, W.C.; Harden, B; Mc Call, I. and Gibson, J.I. Monitoring and Managing pH and EC Using the PourThru Extraction Method. Horticulture Information Leaflet 590, 17p.
Rodella, A. A.; Alcarde, J. C. Avaliação de materiais orgânicos empregados como fertilizantes. Scientia Agrícola, Piracicaba, 51 (3): 556-562, 1994
Williams, S. (ed) Official methods of analysis of the Association of Official Analytical Chemists. 14 ed. Arlington: AOAC, 1984. 1141p.
Kamogawa, M. Y.Avaliação de procedimentos de preparo de amostras contendo enxofre elementar e zinco: relatório técnico. Piracicaba: Universidade de São Paulo, 2016. 14p.
Marcel, B.; Yuncong, C. L.; Guodong, L.; Zhenli, H.; et al.Characterizing Polyhalite Plant Nutritional Properties. Agri Res & Tech: Open Access J. 2017; 6(3): 555690. DOI: 10.19080/ARTOAJ.2017.06.555690.
Margato, V.Avaliação de procedimentos de abertura de amostras de polihalita. Uberaba: Margato Planejamento e Consultoria, 2018. 9p.
Margato, V.Teste e comparação de métodos de abertura de amostras de polyhalita. Uberaba: Margato Planejamento e Consultoria, 2018. 10p.
TEIXEIRA, P. C. (ed. téc.). et al.Manual de métodos de análise de solo. 3. ed. rev. e ampl. Brasília, DF: Embrapa, 2017. p. 199.
U.S. EPA. 1996. "Method 3050B (SW-846): Acid Digestion of Sediments, Sludges, and Soils," Revision 2. Washington, DC.
U.S. EPA. 2007. "Method 3051A (SW-846): Microwave Assisted Acid Digestion of Sediments, Sludges, Soils, and Oils," Revision 1. Washington, DC.
U.S. EPA. 1996. "Method 3052 (SW-846): Microwave Assisted Acid Digestion of Siliceous and Organically Based Matrices," Revision 0. Washington, DC.
U.S. EPA. 2007. "Method 7471B (SW-846): Mercury in Solid or Semisolid Waste (Manual Cold Vapor Technique)," Revision 2. Washington, DC.
U.S. EPA. 1998. "Method 7473 (SW-846): Mercury in Solids and Solutions by Thermal Decomposition, Amalgamation, and Atomic Absorption Spectrophotometry," Revision 0. Washington, DC.
ROCHA, F. S. Estudo da metodologia analítica para determinação de cromo em fertilizantes e concentrados de micronutrientes. 2013. 72p. Trabalho de conclusão de curso (Química Agroindustrial) – Instituto Federal de Educação, Ciência e Tecnologia de Goiás, Goiânia, 2013.
ROSA, A. F. Desenvolvimento de metodologia analítica e validação para os elementos Arsênio e Mercúrio em fertilizantes inorgânicos. 2012. 84p. Trabalho de conclusão de curso (Química Agroindustrial) – Instituto Federal de Educação, Ciência e Tecnologia de Goiás, Goiânia, 2012.
OLIVEIRA, C. B. A. Otimização, validação e estudo comparativo de dois métodos de digestão para determinação de mercúrio em insumos agrícolas orgânicos. 2014. 80f. Dissertação (Mestrado) – Mestrado em Tecnologia de Processos Sustentáveis, Coordenação do Programa de Mestrado em Tecnologia de Processos Sustentáveis - Instituto Federal de Educação, Ciência e Tecnologia de Goiás, Goiânia, 2014.
BRASIL, E. P. F. et al. Chemical Extractors to Assess Potassium Availability in Glauconitic Siltstone. Journal of Agricultural Science, v. 12, n. 9, 2020.
MINISTÉRIO DA AGRICULTURA E PECUÁRIA (Brasil). Instrução Normativa SDA nº 17, de 21 de maio de 2007 alterada pela Instrução Normativa SDA nº 31, de 23 de outubro de 2008. Aprova os Métodos Analíticos Oficiais para Análise de Substratos e Condicionadores de Solos, na forma do Anexo à presente Instrução Normativa. Diário Oficial da União, de 24 de outubro de 2008, Seção 1. Brasília, DF.
ISO 20280:2007(en) Soil quality — Determination of arsenic, antimony and selenium in aqua regia soil extracts with electrothermal or hydride-generation atomic absorption spectrometry.
Official Methods of Analysis of AOAC INTERNATIONAL (1980), AOAC INTERNATIONAL, Gaithersburg, MD, USA, Official Method 892.01.
As sugestões para aprimoramento ou possíveis correções deste documento devem ser direcionadas ao Departamento responsável, para alinhamento das melhores práticas de mercado, legislação vigente e/ou regulamentações, que não tenham sido contempladas na versão vigente.