¶ Folha de Rosto
© 2025 Ministério da Agricultura e Pecuária. Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial. A responsabilidade pelos direitos autorais de textos e imagens desta obra é do autor.
Ano 2025
Elaboração, distribuição, informações:
Ministério da Agricultura e Pecuária
Secretaria de Defesa Agropecuária - SDA
Departamento de Serviços Técnicos - DTEC
Esplanada dos Ministérios, Bloco D, Anexo, Ala B, 4º andar, sala 433
CEP: 70043-900, Brasília - DF
www.agricultura.gov.br
e- mail: cgal@agro.gov.br
Central de Relacionamento: 0800 704 1995
Equipe Técnica:
Bruno Parente Lima — LFDA/PE (org.)
Carolina Maso Viegas - LFDA/RS
Érico Silva Pires — LFDA/GO
Heitor Daguer — DIDEL/COSLAB/CGAL
Juliana Ferreira Ladeira — DICRED/COSLAB/CGAL
Livia Cavaletti Corrêa da Silva — LFDA/SP
Luís Henrique Simões — LFDA/SP
Marcelo Farias de Andrade — LFDA/PE
Michele Fabiane de Oliveira — LFDA/MG
Renan de Almeida Maciel — LFDA/PA
Ricardo Pimenta — SLAV/LFDA/RS
¶ Folha de resumo
| Macroprocesso: Laboratórios |
Objetivo: Estabelecer os métodos oficiais de ensaios para análise de amostras de produtos de origem animal destinados aos laboratórios oficiais e credenciados da Rede Nacional de Laboratórios Agropecuários. |
|
| Processo: Análises Laboratoriais |
||
| Entrega: Segurança e qualidade de alimentos |
Público alvo e demais interessados: Laboratórios oficiais ou credenciados do Ministério da Agricultura e Pecuária (Mapa) |
Versão do documento: 8ª ed. |
| Setor responsável e responsabilidades A Coordenação-Geral de Laboratórios Agropecuários do Departamento de Serviços Técnicos é responsável pela elaboração, atualização e envio para aprovação deste manual, tendo responsabilidade quanto aos procedimentos descritos no documento. |
||
¶ Carnes e produtos cárneos
¶ Preparo de amostras
Instruções específicas, requeridas e descritas nos métodos, também devem ser observadas. Caso a análise não se inicie imediatamente, manter a amostra preparada sob refrigeração em recipiente hermeticamente fechado. Para prevenir perda de água durante o preparo, utilizar porções tão grandes quanto possível.
¶ Carnes in natura e produtos cárneos secos, curados ou defumados
Em moinho: Moer rapidamente por três vezes em moedor com abertura de 3 mm, misturando após cada moagem.
Em processador de alimentos: Cortar a amostra em pedaços de aproximadamente 5 cm e transferir, juntamente com qualquer líquido, para o recipiente do equipamento. Processar por 30 s, raspar as laterais do recipiente com espátula e reincorporar à porção principal da amostra, assim como qualquer material preso nas lâminas. Processar por 30 s adicionais. Repetir este processo por duas vezes. Equipamentos que realizem a raspagem simultaneamente ao processamento podem efetuar o preparo em uma única etapa de aproximadamente 1 min.
Para análise de conservantes permitidos apenas como tratamento de superfície, retirar o produto da embalagem original e cortar toda a porção superficial, de forma que as partes obtidas tenham profundidade de aproximadamente 5 mm. Se necessário reportar o resultado do ensaio por área, cortar as porções obtidas em quadrados de áreas de aproximadamente 0.2 dm². Com um paquímetro, determinar a área superficial (em dm²) de cada porção. Em seguida, pesar cada porção, obtendo a massa (em kg). Moer e analisar individualmente as porções superficiais previamente medidas e pesadas. Limpar todos os utensílios que entraram em contato com a porção superficial do embutido usando etanol e água quente. Amostras de produtos fatiados devem ser preparadas em sua totalidade, sem necessidade de separação da porção superficial e da porção interna.
¶ Produtos congelados
Se necessário o descongelamento da amostra para o seu preparo, dever-se-á processá-la em sua totalidade, agregando à mesma todo o líquido exsudado.
¶ Produtos enlatados
Preparar todo o conteúdo da lata como em Carnes in natura e produtos cárneos secos, curados ou defumados.
¶ Conservas
Remover o líquido da conserva e preparar todo o conteúdo do recipiente como em Carnes in natura e produtos cárneos secos, curados ou defumados.
¶ Embutidos
Remover os envoltórios não comestíveis e preparar como em Carnes in natura e produtos cárneos secos, curados ou defumados. Para análise de conservantes permitidos apenas como tratamento de superfície, retirar o produto da embalagem original e cortar toda a porção superficial do embutido (envoltório), de forma que as partes obtidas tenham profundidade de aproximadamente 5 mm. Se necessário reportar o resultado do ensaio por área, cortar as porções obtidas em quadrados de áreas de aproximadamente 0,2 dm². Com um paquímetro, determinar a área superficial (em dm²) de cada porção. Em seguida, pesar cada porção, obtendo a massa (em kg). Moer e analisar individualmente as porções superficiais previamente medidas e pesadas. Limpar todos os utensílios que entraram em contato com a porção superficial do embutido usando etanol e água quente.
¶ Produtos salgados
Remover o sal aderido à superfície e preparar como em Carnes in natura e produtos cárneos secos, curados ou defumados.
¶ Gelatina
Se em folhas, quebrar em pequenos pedaços e homogeneizar. Demais apresentações não necessitam de tratamento adicional.
¶ Carne mecanicamente separada
Homogeneizar e analisar como recebido. Para o ensaio “Índice de peróxidos”, separar uma porção da amostra e secar sob vácuo a temperatura inferior a 60°C ou evaporar em banho-maria adicionando duas a três porções de etanol (C₂H₅OH, CAS 64-17-5). Extrair a gordura com éter de petróleo e deixar o éter evaporar espontaneamente, removendo os últimos traços sob aquecimento rápido no banho-maria. Não aquecer excessivamente a gordura extraída, evitando-se sua oxidação. Utilizar a gordura para a determinação do Índice de Peróxidos.
¶ Bibliografia
AOAC International. Official Methods of Analysis of AOAC International, Official Method 935.46. 21 ed. Rockville: 2019.
AOAC International. Official Methods of Analysis of AOAC International, Official Method 983.18. 21 ed. Rockville: 2019.
¶ Ácido benzóico e/ou benzoatos
Utilizar o método descrito na norma NMKL 124, expressando o resultado obtido em mg·kg⁻¹, como um número inteiro. Sempre que a legislação permitir a adição de conservantes apenas na parte externa para tratamento de superfície, analisar a parte interna (massa) e o envoltório separadamente e reportar ambos os resultados. Para produtos fatiados, analisar a totalidade da amostra, reportando apenas um resultado.
-
Resultados inferiores a 10 mg·kg⁻¹ devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores a 10 mg·kg⁻¹ e inferiores a 20 mg·kg⁻¹ devem ser expressos como “detectado”.
Caso a presença de ácido benzoico e/ou benzoato não seja permitida no produto, a curva de calibração deve contemplar um ponto na concentração do limite de detecção do método.
¶ Ácido sórbico e/ou sorbatos
Utilizar o método descrito na norma NMKL 124, expressando o resultado obtido em mg·kg⁻¹, como um número inteiro. Sempre que a legislação permitir a adição de conservantes apenas na parte externa para tratamento de superfície, analisar a parte interna (massa) e o envoltório separadamente e reportar ambos os resultados. Para produtos fatiados, analisar a totalidade da amostra, reportando apenas um resultado.
-
Resultados inferiores a 10 mg·kg⁻¹ devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores a 10 mg·kg⁻¹ e inferiores a 20 mg·kg⁻¹ devem ser expressos como “detectado”.
Caso a presença de ácido sórbico e/ou sorbato não seja permitida no produto, a curva de calibração deve contemplar um ponto na concentração do limite de detecção do método.
¶ Amido - qualitativo
¶ Princípio
A interação entre o íon triiodeto (I₃⁻) e a amilose presente nos amidos forma um composto de forte coloração azul, tendendo para o preto de acordo com a concentração dos reagentes empregados.
¶ Campo de aplicação
Produtos cárneos.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Béquer de 150 mL;
-
Pipetas graduadas de 1 mL e 20 mL;
-
Placa aquecedora;
-
Processador de alimentos ou moinho;
-
Proveta de 50 mL;
-
Tubo de ensaio de 25 mL.
¶ Reagentes e soluções
-
Solução de Lugol:
Dissolver 1,25 g de iodo (I₂, CAS 7553-56-2) e 2,5 g de iodeto de potássio (KI, CAS 7681-11-0) em uma pequena porção de água e diluir para 25 mL.
¶ Preparo da amostra
Cortar a amostra em pedaços (exceto o invólucro, quando presente em produtos embutidos), passar em moinho ou processador de alimentos até obtenção de uma massa homogênea. Em béquer de 150 mL, colocar entre 5 g e 6 g de amostra, adicionar 30 mL de água e aquecer em placa aquecedora até fervura por 5 min. Filtrar, esfriar e transferir uma alíquota de aproximadamente 15 mL do filtrado para o tubo de ensaio.
¶ Procedimento de análise
Adicionar duas a três gotas de solução de Lugol à amostra preparada e observar imediatamente a presença ou ausência de coloração azul. Caso observe-se um coloração enegrecida, repetir o ensaio com uma quantidade menor de Lugol. A coloração pode esvanecer na presença de luz.
¶ Expressão dos resultados
Reportar positivo caso se observe o aparecimento de coloração entre azul acinzentado e azul ou verde, tanto mais escura quanto maior a concentração de amido na amostra. A cor pode esvanecer com o tempo.
¶ Bibliografia
AOAC International. Official Methods of Analysis of AOAC International, Official Method 935.49. 20 ed. Rockville: 2016.
¶ Amido - quantitativo
Utilizar o método descrito na Seção Amido e carboidratos totais ou nas normas ISO 5554 ou ISO 10520, expressando o resultado obtido em “g/100 g” com uma casa decimal.
¶ Amido e carboidratos totais
¶ Princípio
Baseia-se na hidrólise ácida do amido e outros açúcares da amostra, produzindo glicose que depois é convertida em furfurais, os quais reagem com a antrona formando um complexo de coloração esverdeada. A quantificação é feita espectrofotometricamente em 620 nm.
¶ Campo de aplicação
Derivados de pescado, carnes e produtos cárneos.
¶ Materiais e equipamentos
-
Agitador vórtex;
-
Balança analítica com resolução mínima de 0,0001 g;
-
Banho-maria;
-
Balões volumétricos de 250 mL e 500 mL;
-
Centrífuga capaz de gerar uma RCF de 1000×g ou superior;
-
Cubeta de vidro ou quartzo;
-
Espectrofotômetro UV/Vis;
-
Estufa;
-
Micropipetas de 50 μL a 2000 μL;
-
Pipetas volumétricas de 2 mL e 10 mL;
-
Processador de alimentos ou moinho;
-
Tubo de centrífuga de fundo cônico de 15 mL, 25 mL ou 50 mL;
-
Tubos de ensaio.
¶ Reagentes e soluções
-
Solução de ácido sulfúrico 76:24 (v/v):
Adicionar a 760 mL de ácido sulfúrico (H₂SO₄, CAS 7664-93-9) a 98%, 240 mL de água Tipo I.
-
Solução de ácido sulfúrico 0,25 mol·L⁻¹;
-
Solução de etanol (C₂H₅OH, CAS 64-17-5) a 80% (v/v);
-
Éter etílico (C₄H₁₀O, CAS 60-28-7);
-
Solução de antrona (C₁₄H₁₀O, CAS 90-44-8):
Dissolver 0,1 g de antrona em 100 mL de solução de ácido sulfúrico 76:24 (v/v). A solução é estável a 4°C, devendo ser descartada quando se tornar verde. Uma vez que a antrona reage com celulose e outros contaminantes, é essencial lavar com etanol todos os frascos de vidro que serão postos em contato com ela.
-
Solução de D-glicose (C₆H₁₂O₆, CAS 50-99-7) 0,01%:
Esta solução deve ser preparada diariamente por diluição de solução estoque a 1% (m/v) de D-glicose. A solução estoque pode ser mantida refrigerada por até uma semana.
¶ Preparo da amostra
-
Produtos de salsicharia:
Retirar os invólucros, cortar em pedaços, passar em moinho ou processador até obtenção de uma massa homogênea.
-
Carne de aves, bovina e suína:
Retirar porções de várias regiões da amostra. Cortar em pedaços menores e homogenizar em moinho ou processador.
¶ Procedimento de análise
-
Preparo da curva de calibração:
Pipetar alíquotas de 250 μL, 500 μL, 1000 μL, 1500 μL e 2000 μL da solução de D-glicose a 0,01% para tubo de ensaio e adicionar água Tipo I de modo que todos eles venham a conter um volume final de 2 mL, obtendo-se assim as soluções padrões. Seguir conforme o item 11 do procedimento. Zerar o equipamento com o branco de reagentes utilizando 2 mL de água Tipo I. Deve-se utilizar uma curva de calibração recém preparada a cada nova batelada de amostras.
-
Para análise de carboidratos totais e amido, pesar porções independentes de aproximadamente 0,5 g de amostra perfeitamente homogeneizada, diretamente em tubo de centrífuga.
-
Lavar com 5 mL de éter etílico, agitar em vórtex e centrifugar por 5 min, retendo o resíduo centrifugado. Repetir este procedimento por mais duas vezes;
-
Na amostra destinada à análise de amido, lavar o resíduo centrifugado da etapa anterior com 5 mL de solução a 80% (v/v) de etanol a quente (entre 60°C a 70°C), agitar em vórtex e centrifugar por 5 min, retendo o resíduo centrifugado. Repetir este procedimento duas vezes mais;
-
Secar o resíduo centrifugado em estufa a (80 ± 2)°C por no mínimo 1 h;
-
Adicionar 10 mL de solução de ácido sulfúrico 0,25 mol·L⁻¹ e colocar o tubo em banho-maria em temperatura igual ou superior a 90°C, tendo o cuidado de manter o nível da solução contida no tubo abaixo do nível de água do banho;
-
Aquecer por 1 h, mantendo o nível da água do banho acima do nível da solução no tubo, agitando o conteúdo do tubo ocasionalmente;
-
Decorrido o tempo estabelecido, transferir quantitativamente o conteúdo do tubo para balão volumétrico de 500 mL e completar o volume com água. Para amostras com limite legal igual ou inferior a 4% de amido e/ou carboidratos totais, transferir quantitativamente o conteúdo do tubo para balão volumétrico de 250 mL;
-
Homogeneizar e deixar decantar;
-
Pipetar 2 mL do sobrenadante para tubo de ensaio previamente lavado com etanol (a presença de contaminantes ou poeira no tubo pode produzir resultados errôneos);
Obs. Para amostras que contenham acima de 10% de carboidratos totais ou amido, efetuar a diluição apropriada.
-
Adicionar 10 mL de solução de antrona ao tubo referente à amostra e tubos da curva padrão, levando-os ao banho-maria com água em ebulição por 10 min;
-
Retirar do banho e deixar esfriar;
-
Zerar o equipamento com o branco de reagentes e proceder à leitura da absorbância em comprimento de onda de 620 nm;
-
Construir a curva de calibração lançando no eixo das ordenadas os valores de absorbância e no eixo das abscissas as concentrações finais de glicose em 25 μg/2 mL, 50 μg/2 mL, 100 μg/2 mL, 150 μg/2 mL e 200 μg/2 mL.
¶ Cálculo e expressão dos resultados
Para cálculo de amido:
Para cálculo de carboidratos totais:
onde:
C = Concentração na alíquota, obtida da curva padrão, em μg de glicose/2 mL;
D = volume do balão volumétrico utilizado na diluição final (250 ou 500 mL), em mL;
m = massa da amostra, em g;
V = volume da alíquota tomada do balão de 250 mL ou 500 mL, em mL;
0,9 = fator de conversão glicose/amido;
10.000 = fator de conversão μg·g⁻¹ para g/100 g.
Expressar os resultados obtidos em “g/100 g” com uma casa decimal.
¶ Bibliografia
BRASIL. Ministério da Agricultura, Pecuária e Abastecimento. Instrução Normativa nº 20, de 21 de julho de 1999: Oficializa os Métodos Analíticos Físico-Químicos, para Controle de Produtos Cárneos e seus Ingredientes - Sal e Salmoura. Diário Oficial [da] Republica Federativa do Brasil, Brasília, DF, 27 de julho de 1999. Seção 1, p. 10.
ESPAÑA. Ministério de Agricultura y Alimentación. Secretaria General de Alimentación. Métodos oficiales de analysis. v.4, 1990. p. 293-295.
Moraes, O.M.G. e Chaves, M.B. Método espectrofotométrico para determinação de amido em produtos cárneos. In: ENCONTRO NACIONAL DE ANALISTAS DE ALIMENTOS, 1988, Belo Horizonte. Resumos. Belo Horizonte, 1988.
¶ Anidrido sulfuroso e sulfitos
Utilizando o método descrito na norma AOAC 990.28, determinar o teor de sulfitos na amostra, expressando o resultado em “mg de /kg” como um número inteiro.
¶ Atividade de água
Determinar, utilizando um dos métodos descritos na norma ISO 18787 e em acordo com as instruções do fabricante do instrumento utilizado, o valor da atividade de água, reportando o resultado obtido com duas casas decimais.
¶ Cálcio em base seca
Determinar o teor de cálcio na amostra utilizando o método descrito nas normas AOAC 983.19, AOAC 2011.14 ou NMKL 153. Com o auxílio do teor de umidade na amostra, determinado conforme este manual, expressar o teor de cálcio em base seca em “g/100 g de base seca” com uma casa decimal:
onde:
C = Teor de cálcio na amostra, em g/100 g;
U = Teor de umidade na amostra, em g/100 g.
¶ Cloreto de sódio (NaCl)
Determinar o teor de cloreto de sódio, utilizando o método descrito na Seção Pescados e subprodutos da pesca - Cloreto de Sódio (NaCl). Reportar o resultado obtido em “g de NaCl/100 g” com uma casa decimal.
¶ Colágeno
Determinar a proporção de colágeno em relação ao teor total de proteína bruta de acordo com o método AOAC 990.26 ou NMKL 127, expressando o resultado em “%” com uma casa decimal.
¶ Corantes artificiais
Utilizando uma das metodologias descritas na norma ISO 13496, reportar “presença de nome comum (código INS)” para cada corante identificado. Reportar “presença de corante hidrossolúvel não identificado” caso seja detectado um corante sem correlação com os corantes testados no método.
¶ Detecção de formaldeído
Utilizar o método B da norma AOAC 931.08. Reportar “positivo” caso seja detectada a presença de formaldeído na amostra e “negativo” em caso contrário.
¶ Detecção de polifosfatos
Utilizar o método descrito na Seção Pescados e subprodutos da pesca - Detecção de polifosfatos.
¶ Detecção de tecidos não permitidos por microscopia
¶ Princípio
Este método tem por objetivo a detecção visual de tecidos inferiores (ossos, tendões e aponeuroses, coágulos, cartilagens, linfonodos, entre outros) em amostras de carne moída por observação direta e com auxílio de microscópio estereoscópio para confirmação.
¶ Campo de aplicação
Carne moída.
¶ Materiais e equipamentos
-
Bandejas;
-
Estiletes;
-
Luminária;
-
Microscópio estereoscópio, com aumento mínimo de 40 vezes;
-
Placas de Petri;
-
Pinças pontiagudas;
-
Tábuas para corte de carne;
¶ Reagentes e soluções
Não aplicável.
¶ Preparo da amostra
Homogeneizar a amostra e separar aproximadamente 100 g de carne moída em uma bandeja.
¶ Procedimento de análise
-
Análise macroscópica: espalhar na bandeja a amostra homogeneizada formando uma camada fina. Analisar meticulosamente, sob iluminação direta, toda a porção por meio da visualização e do tato objetivando encontrar tecidos inferiores. Os tecidos suspeitos devem ser separados em placa de Petri para, caso necessário, posterior confirmação através de microscopia.
-
Análise microscópica: levar ao microscópio estereoscópio as porções contidas nas placas de Petri. Com auxílio de pinça, separar o fragmento e identificá-lo com base no seu tamanho, forma, cor, textura, dureza, brilho e outras características morfológicas, comparando-os com as amostras de referência caso necessário.
¶ Expressão dos resultados
Quando não são detectados tecidos inferiores, os resultados são expressos como “Não detectados”. Quando são detectados tecidos inferiores, os resultados são expressos como “Detectados”, citando os achados. Em ambos os casos, devem ser citados os limites de detecção obtidos em procedimentos de validação para os analitos pesquisados.
¶ Bibliografia
Brasil. Agência Nacional de Vigilância Sanitária. Resolução-RDC Nº 14, de 28/03/2014. Dispõe sobre matérias estranhas macroscópicas e microscópicas em alimentos e bebidas, seus limites de tolerância e dá outras providências. Brasília: Diário Oficial União, 31/03/2014.
Brasil. Ministério da Agricultura, Pecuária e Abastecimento. Instrução Normativa MAPA nº 83, de 21/11/2003. Regulamento técnico de identidade e qualidade de carne bovina em conserva (corned beef) e carne moída de bovino. Brasília: Diário Oficial União: 03/12/2003.
EURACHEM. The fitness for purpose of analytical methods: a laboratory guide to method validation and related topics. 61 p. 1998.
¶ Determinação da Relação U/P em aves
¶ Princípio
Fundamenta-se na determinação do teor de umidade, proteína e sua relação em cortes de aves resfriados ou congelados e carcaças de aves resfriadas, incluindo-se líquidos agregados à embalagem do produto.
¶ Campo de aplicação
Cortes de aves resfriados ou congelados e carcaças de aves resfriadas.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Moinho ou processador para triturar e homogenizar a amostra até obtenção de uma massa homogênea, capaz de homogeneizar amostras com osso;
-
Papel toalha;
-
Sacos plásticos impermeáveis, com capacidade mínima de 4 L.
¶ Reagentes e soluções
Não aplicável.
¶ Preparo da amostra
Manter as amostras sob refrigeração ou congelamento, de acordo com sua exigência de armazenamento.
¶ Procedimento de análise
O ensaio consiste na análise de três amostras individuais (n = 3), não sendo permitida a formação de uma única amostra analítica a partir das três unidades amostrais. Para cada unidade amostral:
-
Verificar se a embalagem primária está intacta, não realizando a análise caso a mesma esteja danificada;
-
Limpar e enxugar o exterior da embalagem;
-
Pesar o produto em sua embalagem original e obter a massa (m₀);
-
Pesar um saco plástico impermeável (m₁);
-
Abrir a embalagem, transferir a amostra para o saco plástico impermeável, tomando cuidado para que não haja perda de amostra, líquido ou gelo. Pesar o conjunto (m₂);
-
Secar a embalagem original do produto e pesar (m₃);
-
Transferir o conteúdo do saco plástico (item e) para o moinho/processador e triturar até obter uma massa homogênea;
-
Determinar o teor de umidade (%U) da amostra de acordo com a norma ISO 1442 ou AOAC 2008.06;
-
Determinar o teor de proteína (%P) da amostra de acordo com a norma ISO 1871 ou AOAC 992.15, utilizando fator de conversão de nitrogênio para proteína de 6,25.
¶ Cálculo e expressão dos resultados
Determinar a massa do líquido residual na embalagem (Ml) de cada amostra, em gramas:
Calcular o percentual total de umidade nas amostras, %Ut, em “g/100 g”:
Calcular o percentual total de proteína nas amostras, %Pt, em “g/100 g”:
Calcular a relação umidade/proteína das amostras (U/P):
Reportar a média dos valores da Relação U/P das três amostras com duas casas decimais.
¶ Bibliografia
COMUNIDADE EUROPEIA. Regulamento (CE) N.º543/2008 da Comissão de 16 de junho de 2008. Estabelece regras de execução do Regulamento (CE) n.º1234/2007. Jornal Oficial da União Europeia, [s.l.]:2008.
¶ Determinação de conservantes por cromatografia líquida acoplada à espectrometria de massas
¶ Princípio
O método se baseia na extração sólido-líquido com partição em baixa temperatura, utilizando mistura de solventes orgânicos com baixa proporção de água e acidificados com ácido acético. A separação dos analitos é realizada através da cromatografia líquida em fase reversa utilizando fase estacionária cianopropil. A quantificação é realizada por espectrometria de massas, utilizando fonte de ionização por electrospray e padronização interna. O escopo do método abrange os analitos: ácido benzoico (BEN), ácido sórbico (SOR), butilparabeno (BuP), etilparabeno (EtP), hexametilenotetramina (HMT), metilparabeno (MtP), natamicina (NAT), nisina (NIS) e propilparabeno (PrP).
¶ Campo de aplicação
Produtos cárneos e pescados.
¶ Materiais e equipamentos
-
Agitador de tubos tipo vortex;
-
Agitador orbital;
-
Balança analítica;
-
Balões volumétricos de 10 mL;
-
Banho de ultrassom;
-
Centrífuga refrigerada para tubos de 15 mL e 50 mL;
-
Centrífuga refrigerada para microtubos;
-
Coluna para cromatografia líquida com fase estacionária di-isopropril-3-cianopropil silano ligado à sílica hidroxilada, com partículas de diâmetro nominal de no mínimo 3,5 μm (Zorbax-Agilent SB-CN, ou similar);
-
Dispensador automático (volume mínimo 10 mL);
-
Homogeneizador/disruptor de amostras (Ultra Turrax ou similar);
-
Micropipetadores e ponteiras para volumes de 2-20 μL; 10-100 μL; 100-1000 μL e 5000-10000 μL;
-
Microtubos de polipropileno, com capacidade para 1,5 mL;
-
Provetas de 250 mL e 500 mL;
-
Sistema de cromatografia líquida acoplada a espectrômetro de massas em modo tandem, com fonte de ionização por electrospray (LC-ESI-MS/MS);
-
Tubos de polipropileno, com capacidade para 15 mL e 50 mL (tipo falcon);
-
Vials de borossilicato para HPLC com capacidade de 1,5 mL.
¶ Reagentes e soluções
-
Solução extratora de acetonitrila (CH₃CN, CAS 75-05-8) : metanol (CH₃OH, CAS 67-56-1) : água (45:45:10 v/v/v) acidificada com 0,1% ácido acético (CH3COOH, CAS 64-19-7).
-
Fase móvel A (solução aquosa de ácido fórmico (HCOOH, CAS 64-18-6) 0,1%).
-
Fase móvel B (solução de acetonitrila acidificada com 0,1% de ácido fórmico).
-
Fase móvel de diluição (fase móvel A:fase móvel B 90:10 v/v).
-
Soluções-estoque de conservantes: nisina A (NIS,C₁₄₃H₂₃₀N₄₂O₃₇S₇, CAS 141-45-5) a 1 g·L⁻¹, preparada em água:acetonitrila (50:50 v/v), acidificada com ácido fórmico em 0,1%; ácido benzoico (BEN, C₆H₅COOH, CAS 65-85-0) a 10 g·L⁻¹, preparada em metanol com baixa proporção de água para dissolução; hexametilenotetramina (HMT, C₆H₁₂N₄, CAS 100-97-0) a 10 g·L⁻¹, ácido sórbico (SOR, C₅H₇COOH, CAS 110-44-1) a 1 g·L⁻¹, natamicina (NAT, C₃₃H₄₇NO₁₃, CAS 7681-93-8) a 1 g·L⁻¹, metilparabeno (MtP, C₈H₈O₃,CAS 99-76-3) a 1 g·L⁻¹, etilparabeno (EtP, C₉H₁₀O₃, CAS 120-47-8) a 1 g·L⁻¹; propilparabeno (PrP, C₁₀H₁₂O₃, CAS 94-13-3) a 1 g·L⁻¹ e butilparabeno (BuP, C₁₁H₁₄O₃, CAS 94-26-8) a 1 g·L⁻¹, todos preparados em metanol.
-
Soluções-estoque do padrão interno ácido 3,5-dinitrobenzóico (3,5-DNB, C₇H₄O₆N₂, CAS 99-34-3) a 1 g·L⁻¹, preparada em metanol.
-
Solução-mix de fortificação do padrão interno 3,5-DNB (10 mg·L⁻¹), preparada em metanol.
-
Solução-mix de fortificação de conservantes, preparada em metanol de forma que a concentração final de cada analito seja: 50 mg·L⁻¹ para MtP, EtP, PrP, BuP e NAT; 100 mg·L⁻¹ para NIS e SOR e 1000 mg·L⁻¹ para BEN.
Os solventes orgânicos devem ser de grau LC-MS e a água deve ser de grau ultrapuro. Utilizar banho de ultrassom para dissolução dos padrões. As soluções de fortificação preparadas conforme este método são estáveis por no mínimo 30 dias quando estocadas a (−30 ± 10)°C.
¶ Preparo da amostra
-
Produtos cárneos salgados, secos curados e/ou maturados (não embutidos):
As amostras devem ser preparadas em duas porções, uma contendo somente a superfície e outra contendo a porção interna do produto.
-
Produtos cárneos secos curados e/ou maturados embutidos:
Retirar os envoltórios, que serão processados para análise. Para os produtos em que são permitidos conservantes apenas na superfície, preparar conforme Seção Carnes in natura e produtos cárneos secos, curados ou defumados. As demais amostras devem ser processadas homogeneizando-se todo o seu conteúdo (incluindo amostras de produtos fatiados).
-
Pescado in natura e salgado:
Limpar o pescado retirando a pele e escamas, espinha, gordura e vísceras. Retirar porções da musculatura de várias regiões do pescado e cortar as amostras em pedaços menores para processar e homogeneizar em processador.
-
Conservas de pescado:
Processar e homogeneizar todo o conteúdo da lata (líquido + parte sólida) em processador.
¶ Procedimento de análise
-
Utilizando balança analítica, pesar (2,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL.
-
Fortificar as amostras com 200 μL do mix de padrão interno.
-
Adicionar 10 mL de solução de extração aos tubos com amostra.
-
Homogeneizar o conteúdo de cada tubo por volta de cinco segundos no homogeneizador/disruptor.
-
Agitar em mesa agitadora orbital durante 20 min.
-
Centrifugar por 10 min à rotação de 3488 g, sob refrigeração.
-
Transferir o sobrenadante para um tubo de polipropileno de 15 mL de capacidade e levar ao freezer (à temperatura inferior a −18°C) por no mínimo 1 h.
-
Centrifugar novamente por 10 min à rotação de 3488 g, sob refrigeração.
-
Transferir uma alíquota (μl) do sobrenadante do extrato para microtubos de polipropileno 1,5 mL e diluir de acordo com a sensibilidade do sistema LC-ESI-MS/MS, utilizando fase móvel de diluição. Homogeneizar e centrifugar a 13.000 g, sob refrigeração. Por fim, transferir o sobrenadante límpido para vials e injetar no sistema LC-ESI-MS/MS.
-
Preparar uma curva analítica em matriz, pesando-se (2,0 ± 0,1) g de amostra “branca” para cada ponto e fortificando as amostras com 200 μL do mix de padrões internos. Estabelecer um gradiente de concentração de acordo com as faixas de trabalho: BEN, HMT, 25 a 200 mg·kg⁻¹; NAT, BuP, EtP, MtP e PrP, 1,25 a 10,0 mg·kg⁻¹; SOR e NIS, 2,5 a 20,0 mg·kg⁻¹. Para isso, adicionar volumes crescentes de 50 μL, 100 μL, 200 μL, 300 μL e 400 μL de cada solução-mix de fortificação e completar o volume com solução de extração q.s.p. 10 mL. Amostras, brancos analíticos, curvas analíticas e demais controles devem estar sujeitos aos mesmos procedimentos analíticos descritos.
-
Parâmetros instrumentais sugeridos:
-
temperatura do forno de coluna: 40°C;
-
volume de injeção: 10 μL;
-
fluxo da fase móvel: 500 μL min⁻¹;
-
gradiente de eluição: 0-1 min, 90% A; 2-3 min, 80% A; 4 min, 70% A, 5-6 min, 50% A; 7-8 min, 10% A; 9 min, 50% A; 10 min, 90% A; mais 4 min para auto equilíbrio do sistema.
-
| Analitos (sigla) | Padrão interno | Íon Precursor (ESI) m/z | Transição de quantificação m/z (EC, V) |
Transição de confirmação m/z (EC, V) |
DP (V) |
|---|---|---|---|---|---|
| Ácido sórbico (SOR) | 3,5-DNB | 112,9 (+) | 67,0 (19) | 65,0 (25) | 56 |
| Ácido benzoico (BEN) | 3,5-DNB | 122,9 (+) | 79,0 (17) | 51,0 (51) | 36 |
| Hexametilenotetramina (HMT) | 3,5-DNB | 141,1 (+) | 112,0 (11) | 98,0 (21) | 51 |
| Nisina (NIS) | – | 672,1 (5+) | 811,3 (23) | 649,3 (23) | 56 |
| Natamicina (NAT) | 3,5-DNB | 666,2 (+) | 503,2 (17) | 485,2 (21) | 56 |
| Metilparabeno (MtP) | 3,5-DNB | 151,0 (-) | 91,9 (-24) | 135,9 (-18) | -50 |
| Etilparabeno (EtP) | 3,5-DNB | 165,0 (-) | 137,0 (-18) | 93,0 (-26) | -10 |
| Propilparabeno (PrP) | 3,5-DNB | 179,0 (-) | 92,9 (-16) | 108,0 (-30) | -40 |
| Butilparabeno (BuP) | 3,5-DNB | 193,0 (-) | 136,9 (-20) | 92,9 (-26) | -105 |
| 3,5-Ácido dinitrobenzoico (3,5-DNB) | – | 210,9 (-) | 166,9 (-14) | – | -45 |
¶ Expressão dos resultados
A identificação de cada espécie química é feita pelo tempo de retenção (min) e o perfil de fragmentação molecular, através da razão iônica que deve ser de no mínimo 20% entre as transições (quantificação/confirmação). A quantificação é realizada através da padronização interna, utilizando relação funcional linear de razão das concentrações (eixo x) dos analitos versus razão de áreas dos picos (eixo y), exceto para a nisina, determinada por padronização externa. Expressar separadamente o resultado de cada porção analisada da amostra. Expressar os resultados de ácido benzoico e de ácido sórbico conforme os itens Carnes e produtos cárneos - Ácido benzóico e/ou benzoatos e Carnes e produtos cárneos - Ácido sórbico e/ou sorbatos. Os demais conservantes devem ser expressos em “mg/kg” com no máximo quatro algarismos significativos. Para os produtos em que o uso de natamicina é permitido para tratamento de superfície, os resultados da parte externa devem ser expressos em “mg/dm²”.
¶ Bibliografia
Luciano Molognoni, Heitor Daguer, Leandro Antunes de Sá Ploêncio, Juliano de Dea Lindner. A multi-purpose tool for food inspection: simultaneous determination of various classes of preservatives and biogenic amines in meat and fish products by LC-MS. Talanta, vol. 178, p. 1053-1066, 2018.
¶ Índice de peróxidos
Utilizar o método descrito na norma ISO 3960 para determinação do índice de peróxidos na porção gordurosa da amostra, expressando o resultado final em “meq de /kg de gordura” com uma casa decimal.
¶ Lipídios totais
Determinar o teor de lipídios totais utilizando o método descrito na norma ISO 1443 ou, opcionalmente, na norma NMKL 181 ou na norma AOAC 2008.06. Reportar o valor obtido em “g/100 g” com uma casa decimal.
¶ Nitritos e nitratos
Determinar o teor de nitritos e nitratos em “mg de NaNO₂/kg” utilizando o método descrito nas normas NMKL 165 (nitritos e nitratos), NMKL 194 (nitritos e nitratos) ou ISO 2918 (nitritos) e ISO 3091 (nitratos) ou, alternativamente, o método descrito na Seção Nitritos e nitratos por eletroforese capilar de zona. Reportar o resultado conforme descrito abaixo:
Nitritos
-
Resultados inferiores a 10 mg de NaNO₂/kg devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores a 10 mg de NaNO₂/kg e inferiores a 20 mg de NaNO₂/kg devem ser expressos como “detectado”;
-
Resultados maiores ou iguais a 20 mg de NaNO₂/kg devem ser expressos arredondados para um número inteiro.
Nitratos
-
Resultados inferiores a 10 mg de NaNO₂/kg devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores 10 mg de NaNO₂/kg e inferiores a 30 mg de NaNO₂/kg devem ser expressos como “detectado”;
-
Resultados maiores ou iguais a 30 mg de NaNO₂/kg devem ser expressos arredondados para um número inteiro.
Uma nova curva de calibração deve ser obtida a cada batelada de análises, independentemente do método utilizado.
¶ Nitritos e nitratos por eletroforese capilar de zona
¶ Princípio
O método se baseia na extração dos analitos com água quente e tetraborato de sódio. A separação dos analitos é realizada através de eletroforese capilar de zona, com detecção por arranjo de diodos (CZE-DAD). A quantificação é feita por padronização interna com tiocianato de potássio.
¶ Campo de aplicação
Carnes, pescados e produtos derivados.
¶ Materiais e equipamentos
-
Agitador de tubos (tipo vórtex);
-
Balança analítica;
-
Banho-maria com agitação;
-
Centrífuga refrigerada para tubos de 15 mL e 50 mL;
-
Centrífuga refrigerada para microtubos;
-
Capilares de sílica fundida, com 75 μm de diâmetro interno e 48,5 cm de comprimento total;
-
Funis;
-
Homogeneizador/disruptor de amostras (Ultra Turrax ou similar);
-
Membrana filtrante de polietileno e poros com diâmetro de 0,45 μm (ou menor);
-
Micropipetadores e ponteiras para volumes de 1000-10000 μL, 100-1000 μL e 5-200 μL;
-
Microtubos de polipropileno, com capacidade para 1,5 mL;
-
Papel de filtro qualitativo;
-
Sistema automático de eletroforese capilar, com detector de arranjo de diodos, controlador de temperatura, operado por software de controle e aquisição de dados;
-
Sistema de filtração de solventes;
-
Tubos de polipropileno, com capacidade para 15 mL e 50 mL (tipo falcon);
-
Vials de polipropileno para eletroforese capilar, com tampas de silicone e capacidade de 1,5 mL.
¶ Reagentes e soluções
-
Acetonitrila de grau cromatográfico.
-
Solução de hidróxido de sódio (NaOH, CAS 1310-73-2) 1,0 mol·L⁻¹;
-
Solução de tetraborato de sódio (B₄Na₂O₇, CAS 1330-43-4) 5% (m/v);
-
Solução de β-alanina (C₃H₇NO₂, CAS 107-95-9) 100 mmol·L⁻¹;
-
Solução de ácido perclórico (ClHO₄, CAS 7601-90-3) 100 mmol·L⁻¹;
-
Soluções-estoque de tiocianato (SCN⁻) 10 g·L⁻¹;
-
Soluções-estoque de nitrato (NO₃⁻) 1 g·L⁻¹;
-
Soluções-estoque de nitrito (NO₂⁻) 1 g·L⁻¹;
-
Solução de fortificação de tiocianato (SCN⁻) 100 mg·L⁻¹;
-
Solução de fortificação de nitrato (NO₃⁻) 100 mg·L⁻¹;
-
Solução de fortificação de nitrito (NO₂⁻) 100 mg·L⁻¹;
-
Eletrólito de corrida (pH ≈ 3,8), preparado com β-alanina 100 mmol·L⁻¹ (2600 μL), ácido perclórico 100 mmol·L⁻¹ (1000 μL) e água ultrapura (400 μL). Filtrar a vácuo em membrana filtrante. Transferir para vials.
¶ Preparo da amostra
Processar e homogeneizar a amostra em processador. Quando confeccionados em material não comestível, os envoltórios deverão ser retirados e descartados. Caso contrário, processar todo o conteúdo (amostra e envoltórios). Analisar a amostra em até 24 horas após a homogeneização.
¶ Procedimento
-
Pesar (2,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL. Se necessário, pode-se pesar uma massa menor que 2 g, corrigindo-se o fator de concentração para cálculo dos analitos.
-
Fortificar as amostras com 150 μL da solução-estoque de tiocianato (10000 mg·L⁻¹);
-
Adicionar 1 mL de solução de tetraborato de sódio 5%;
-
Adicionar 9 mL de água ultrapura;
-
Homogeneizar por aproximadamente 1 min em Ultra Turrax.
-
Colocar os tubos em banho-maria à temperatura de aproximadamente 65°C, sob agitação, por 20 min;
-
Centrifugar à temperatura aproximada de 4°C por 10 min a no mínimo 3000 g;
-
Filtrar em papel de filtro qualitativo;
-
Transferir 100 μL do filtrado para um microtubo, adicionar 200 μL de acetonitrila e 200 μL de água ultrapura. Agitar;
-
Centrifugar à temperatura aproximada de 4°C, por 10 min a cerca de 17000 g;
-
Transferir cerca de 450 μL do sobrenadante para vials e injetar no sistema de eletroforese. Caso necessária, a diluição de amostras deve ser feita com solução de padrão interno preparada na mesma concentração do vial de injeção.
-
Preparar uma curva de calibração em solvente, de acordo com as faixas de trabalho: nitrato, 18,75 mg·L⁻¹ a 600 mg·L⁻¹; nitrito, 9,375 mg·L⁻¹ a 300 mg·L⁻¹; tiocianato, 750 mg·L⁻¹. Transferir para vials e injetar no sistema de eletroforese. Preparar também uma amostra de recuperação, fortificada nos limites regulatórios;
-
Parâmetros instrumentais recomendados: injeção hidrodinâmica da amostra pelo outlet (−50 mbar 3 s); comprimento de onda: 210 nm; voltagem de separação: 20 kV; temperatura do capilar: 20°C; condicionamento do capilar: NaOH 1 mol·L⁻¹ (20 min), água ultrapura (20 min) e eletrólito de corrida (20 min).
¶ Cálculo e expressão dos resultados
A identificação de cada espécie química é feita pelo tempo de migração e pelo espectro de absorção. A quantificação é realizada por padronização interna (750 mg·L⁻¹), utilizando relação funcional linear de razão das concentrações (eixo x) dos analitos versus razão de áreas dos picos (eixo y). Expressar o resultado da amostra conforme descrito na Seção Nitritos e nitratos.
¶ Bibliografia
Fabiana Della Betta, Lais Morilla Pereira, Mariana Araújo Siqueira, Andressa Camargo Valese, Heitor Daguer, Roseane Fett Luciano Vitali, Ana Carolina Oliveira Costa. A sub-minute CZE method to determine nitrate and nitrite in meat products: an alternative for routine analysis. Meat Science, vol. 119, p. 62-68, 2016.
¶ Nitrogênio total
Determinar o teor de nitrogênio na amostra de acordo com a norma ISO 1871, expressando o resultado obtido em “g de N/100 g” com duas casas decimais. Alternativamente, utilizar a norma AOAC 992.15.
¶ pH
Determinar o pH no extrato homogeneizado da amostra, conforme metodologia descrita na norma ISO 2917. Reportar o valor obtido com duas casas decimais.
¶ Proteína
Determinar o teor de nitrogênio na amostra de acordo com a norma ISO 1871. Multiplicar o valor obtido por 6,25 para obtenção do teor de proteína. Expressar o resultado obtido em “g/100 g” com duas casas decimais. Alternativamente, utilizar a norma AOAC 992.15.
¶ Relação Umidade/Proteína
A partir dos resultados para o Teor de Umidade e Teor de Proteínas, obtidos utilizando-se os métodos estabelecidos neste manual, calcular a relação U/P, expressando o resultado final com duas casas decimais.
¶ Resíduo mineral fixo (Cinzas)
Determinar o teor de resíduo mineral fixo (cinzas) utilizando o método descrito na norma ISO 936, reportando o resultado obtido em “g/100 g” com uma casa decimal.
¶ Teste de gotejamento (dripping test)
¶ Princípio
Baseia-se na determinação gravimétrica do teor de líquido perdido pelas aves congeladas no degelo em condições padronizadas. O resultado final do ensaio é a média aritmética do percentual de líquido perdido de seis carcaças.
¶ Campo de aplicação
Carcaças de aves congeladas.
¶ Materiais e equipamentos
-
Balança com resolução mínima de 0,1 g;
-
Banho com circulação de água, controlada termostaticamente à temperatura de (42 ± 2)°C, e volume de água não inferior a oito vezes o volume das carcaças a serem controladas;
-
Barbante;
-
Guias ou pesos para manter as carcaças mergulhadas verticalmente;
-
Sacos plásticos, resistentes e impermeáveis com capacidade suficiente para conter a carcaça e permitir um fechamento seguro;
-
Termômetro de superfície ou de infravermelho;
-
Toalhas de tecido ou papel para enxugar a carcaça.
¶ Reagentes e soluções
Não aplicável.
¶ Preparo da amostra
As carcaças não podem sofrer descongelamento desde a coleta até sua recepção no laboratório, devendo ser mantidas em estado físico congelado sólido. A análise será iniciada estando as aves a uma temperatura de (−12 ± 1)°C, medida em sua superfície externa.
¶ Procedimento
-
Enxugar o lado externo da embalagem de modo a eliminar todo o líquido e gelo aderido;
-
Pesar a ave, obtendo-se M₀;
-
Retirar a carcaça congelada de dentro da embalagem (com as vísceras, se presentes), enxugar a embalagem e pesá-la, obtendo-se M₁;
-
Introduzir a carcaça, com os miúdos, num saco plástico com a cavidade abdominal voltada para o fundo do saco plástico e fechá-lo tendo o cuidado de retirar o excesso de ar por meio de pressão manual;
-
Mergulhar completamente no banho a parte do saco que contêm a carcaça e as vísceras, de tal maneira que a água não penetre no interior do mesmo. Os sacos individuais não devem tocar uns nos outros;
-
Repetir este procedimento para as outras cinco carcaças;
-
Manter os sacos no banho de água a (42 ± 2)°C com circulação contínua de acordo com os tempos definidos na Tabela 2.
-
Após o período de imersão, retirar o saco plástico do banho, abrir um orifício na parte inferior de modo que a água liberada pelo descongelamento possa escorrer e deixar à temperatura ambiente entre 18°C e 25°C por no mínimo 60 min;
-
Retirar a carcaça descongelada do saco e as vísceras da cavidade torácica e enxugar a carcaça interna e externamente;
-
Perfurar o invólucro das vísceras, deixar escoar e secar o invólucro e as vísceras descongeladas;
-
Pesar a carcaça descongelada juntamente com as vísceras e seu invólucro, obtendo-se M₂;
-
Pesar o invólucro que continha as vísceras, obtendo-se M₃.
Caso as aves não contenham vísceras, M₂ será o peso da carcaça descongelada e M₃ será igual a zero.
| Massa da ave mais vísceras, em g | Tempo de imersão, em min |
|---|---|
| Até 800 | 65 |
| 801 a 900 | 72 |
| 901 a 1000 | 78 |
| 1001 a 1100 | 85 |
| 1101 a 1200 | 91 |
| 1201 a 1300 | 98 |
| 1301 a 1400 | 105 |
| Acima de 1400 g, aumentar o tempo de permanência no banho em 7 min para cada 100 g adicionais na massa da ave congelada. | |
¶ Cálculo e expressão dos resultados
Reportar o valor do “% de líquido perdido” por carcaça e a média aritmética do “% de líquido perdido” com uma casa decimal.
¶ Bibliografia
Sônia Dedeca da Silva de Campos. Parâmetros de qualidade de frangos congelados: caracterização e alterações decorrentes da estocagem. Tese: Doutorado em Engenharia de Alimentos. Universidade Estadual de Campinas. Campinas: 1993.
FAO. Manual of food quality control 3. Roma: 1979.
¶ Umidade
Determinar o teor de Umidade utilizando o método descrito na norma ISO 1442, reportando o resultado obtido em “g/100 g” com uma casa decimal. Alternativamente, utilizar o método descrito na norma AOAC 2008.06.
¶ Leite e produtos lácteos
¶ Acidez
¶ Caseínas – acidez livre
Utilizar o método descrito na norma IDF 91, expressando o resultado com duas casas decimais em “mL NaOH 0,1 N/g”.
¶ Creme de leite, leite fluido e nata
Utilizar o método descrito na norma AOAC 947.05, expressando o resultado com duas casas decimais em “g de ác. lático/100 g” ou “g de ác. lático/100 mL”, conforme aplicável.
¶ Gordura anidra do leite (butter oil)
Utilizar o método descrito na norma IDF 6, expressando o resultado com duas casas decimais em “g de ác. oleico/100 g de gordura”.
¶ Leite em pó – acidez titulável
Utilizar o método descrito na norma IDF 86, expressando o resultado com uma casa decimal em “mL NaOH 0,1 N/10 g SNG”.
¶ Leites fermentados e sobremesas lácteas fermentadas
Utilizar o método descrito na norma IDF 150, expressando o resultado com duas casas decimais em “g de ác. lático/100 g”.
¶ Manteiga
Utilizar o método descrito na norma IDF 6, expressando o resultado com duas casas decimais em “milimoles/100 g de matéria gorda”.
¶ Acidez em manteiga da terra e manteiga comum
¶ Princípio
Fundamenta-se na reação de neutralização pelo hidróxido de sódio em presença de fenolftaleína como indicador.
¶ Campo de aplicação
Este método é aplicável, unicamente, a manteiga da terra (manteiga de garrafa) e manteiga comum. Este método não tem equivalência com o método descrito na norma IDF 6.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,01 g;
-
Bureta com resolução mínima de 0,05 mL;
-
Centrífuga;
-
Erlenmeyer ou béquer de 100 mL;
-
Estufa;
-
Papel de filtro qualitativo;
-
pH-metro;
-
Proveta de 50 mL.
¶ Reagentes e soluções
-
Solução alcoólica de fenolftaleína (C₂₀H₁₄O₄, CAS 77-09-8) a 1% (m/v);
-
Solução padronizada de hidróxido de sódio (NaOH, CAS 1310-73-2) a 0,1 mol·L⁻¹;
-
Solução de etanol (C₂H₅OH, CAS 64-17-5) e éter etílico (C₄H₁₀O, CAS 60-28-7) (1+2) (v/v).
¶ Preparo da amostra
Transferir para um béquer cerca de 50 g de amostra e fundir em estufa entre 50°C e 60°C. Deixar repousar para separar as camadas. Decantar a camada gordurosa, filtrando através de papel de filtro seco para um béquer também seco. Alternativamente, centrifugar para separação das porções aquosa e gordurosa.
¶ Procedimento de análise
-
Pesar entre 4,90 g e 5,10 g da porção gordurosa em béquer de 100 mL;
-
Adicionar 40 mL da solução álcool-éter;
-
Adicionar 5 gotas de solução alcoólica de fenolftaleína a 1% (m/v), para uma titulação colorimétrica, ou levar ao pH-metro, para uma titulação potenciométrica;
-
Titular a amostra com solução de hidróxido de sódio 0,1 mol·L⁻¹ até aparecimento de coloração rósea persistente por aproximadamente 30 segundos ou pH 8,4;
-
Repetir o procedimento utilizando apenas 40 mL da solução álcool-éter (branco).
¶ Cálculo e expressão dos resultados
onde:
V = volume da solução de hidróxido de sódio 0,1 mol·L⁻¹ gasto na titulação, em mL;
Vbranco = volume da solução de hidróxido de sódio 0,1 mol·L⁻¹ gasto na titulação do branco, em mL;
f = fator de correção da solução de hidróxido de sódio 0,1 mol·L⁻¹;
m = massa da amostra, em gramas;
0,1 = concentração molar da solução de hidróxido de sódio.
Expressar os resultados com duas casas decimais.
¶ Bibliografia
BRASIL. Ministério da Agricultura. Secretaria Nacional de Defesa Agropecuária. Laboratório Nacional de Referência Animal. Métodos analíticos oficiais para controle de produtos de origem animal e seus ingredientes: II – Métodos físicos e químicos. Brasília: 1981.
¶ Ácido benzoico e/ou benzoatos
Utilizar o método descrito na norma NMKL 124, expressando o resultado obtido em mg·kg⁻¹, como um número inteiro.
-
Resultados inferiores a 10 mg·kg⁻¹ devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores a 10 mg·kg⁻¹ e inferiores a 20 mg·kg⁻¹ devem ser expressos como “detectado”.
Caso a presença de ácido benzoico e/ou benzoato não seja permitida no produto, a curva de calibração deve contemplar um ponto na concentração do limite de detecção do método.
Uma nova curva de calibração deve ser obtida a cada batelada de análises.
¶ Ácido sórbico e/ou sorbatos
Utilizar o método descrito na norma IDF 139 ou, alternativamente, na norma NMKL 124, expressando o resultado obtido em mg·kg⁻¹, como um número inteiro.
-
Resultados inferiores a 10 mg·kg⁻¹ devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores a 10 mg·kg⁻¹ e inferiores a 20 mg·kg⁻¹ devem ser expressos como “detectado”.
Caso a presença de ácido sórbico e/ou sorbato não seja permitida no produto, a curva de calibração deve contemplar um ponto na concentração do limite de detecção do método.
Uma nova curva de calibração deve ser obtida a cada batelada de análises.
¶ Açúcares em leite condensado
Determinar o teor de sacarose, glicose ou frutose de acordo com o método descrito na Seção Lactose e sacarose por cromatografia líquida com detecção por índice de refração ou pelo método descrito na Seção Lactose, glicose, frutose e sacarose por cromatografia iônica. Caso o produto apresente apenas os açúcares sacarose e lactose em sua formulação, pode-se utilizar o método descrito na norma IDF 35 para quantificação da sacarose. Reportar os teores dos açúcares quantificados em “g/100 g” com uma casa decimal.
Uma nova curva de calibração deve ser obtida a cada batelada de análises, independentemente do método utilizado.
¶ Amido – qualitativo
¶ Princípio
A interação entre o íon e a amilose presente nos amidos forma um composto de forte coloração azul, tendendo para o preto de acordo com a concentração dos reagentes empregados.
¶ Campo de aplicação
Leite fluido, condensado, fermentado e em pó, doce de leite, queijo, requeijão e ricota.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Béquer de 150 mL;
-
Pipetas graduadas de 1 e 20 mL;
-
Placa aquecedora;
-
Processador de alimentos ou moinho;
-
Proveta de 50 mL;
-
Tubo de ensaio de 25 mL.
¶ Reagentes e soluções
-
Amido solúvel ((C₆H₁₀O₅)ₙ, CAS 9005-84-9);
-
Solução de Lugol:
Dissolver 1,25 g de iodo (I₂, CAS 7553-56-2) e 2,5 g de iodeto de potássio (KI, CAS 7681-11-0) em uma pequena porção de água e diluir para 25 mL.
¶ Preparo da amostra
-
Leite condensado, doce de leite, queijo, requeijão e ricota:
Pesar 10 g da amostra homogeneizada em béquer de 150 mL, adicionar 50 mL de água e misturar. Aquecer em placa aquecedora, mantendo em ebulição por aproximadamente 3 min. Deixar esfriar até temperatura ambiente antes da execução do teste.
-
Leite fluido e leite fermentado:
Homogenizar a amostra a temperatura ambiente, agitando e invertendo o recipiente ou embalagem cinco ou seis vezes. Quando a amostra contiver grumos de creme, aquecer de 38°C a 40°C em banho-maria, esfriar até temperatura ambiente agitando ocasionalmente. Transferir 10 mL da amostra para tubo de ensaio, aquecer até ebulição em banho-maria e deixar por 3 min. Deixar esfriar até temperatura ambiente antes da execução do teste.
-
Leite em pó:
Homogenizar a amostra, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes. Reconstituir de acordo com as instruções do fabricante e transferir 10 mL da amostra para tubo de ensaio, aquecer até ebulição em banho-maria e deixar por 3 min. Deixar esfriar até temperatura ambiente antes da execução do teste.
¶ Procedimento de análise
Adicionar 250 μL de solução de Lugol à amostra preparada e observar a ocorrência da formação de uma coloração azul ou verde. A cor pode esvanecer com o tempo.
¶ Expressão dos resultados
Reportar positivo caso se observe o aparecimento de coloração entre azul acinzentado e azul ou verde.
Para amostras de leite, pode-se utilizar como referência uma amostra de leite sabidamente negativo adicionado de 0,4 g l⁻¹ de amido.
¶ Bibliografia
Carina de Souza Gondim, Roberto Cesar Santos de Souza, Marina de Paula Penna e Palhares, Roberto Gonçalves Junqueira e Scheilla Vitorino Carvalho de Souza. Performance improvement and single laboratory validation of classical qualitative methods for the detection of adulterants in milk: starch, chlorides and sucrose. Analytical Methods, vol. 7(22), p. 9692-9701, 2015.
Carina de Souza Gondim, Roberto Gonçalves Junqueira e Scheilla Vitorino Carvalho de Souza. Interlaboratory validation of modified classical qualitative methods for detection of adulterants in milk: starch, chloride, and sucrose. Food Analytical Methods, vol. 9, p. 2509-2520, 2016.
¶ Cinzas
¶ Caseína alimentar ao ácido e láctica
Utilizar o método descrito na norma IDF 89, expressando o resultado com uma casa decimal em “g/100 g”.
¶ Caseína alimentar ao coalho, caseinatos e soro de leite em pó
Utilizar o método descrito na norma IDF 90, expressando o resultado com uma casa decimal em “g/100 g”.
¶ Doce de leite
Utilizar o método descrito na norma AOAC 930.30, expressando o resultado com uma casa decimal em “g/100 g”.
¶ Leite de cabra
Utilizar o método descrito na norma AOAC 945.46, expressando o resultado com uma casa decimal em “g/100 g”.
¶ Cloreto de sódio em manteiga
Utilizar o método descrito nas normas IDF 12 ou IDF 179, expressando o resultado obtido em “g de /100 g” com duas casas decimais.
Em amostras de manteiga adicionada de cristais de sal, realizar o ensaio em triplicata.
¶ Corantes artificiais
Utilizando a metodologia descrita na norma NMKL 130, expressar o resultado identificando o corante encontrado na forma “nome comum (código INS)” para cada corante identificado, reportando a quantidade encontrada em mg·kg⁻¹ como um número inteiro. Reportar “presença de corante hidrossolúvel não identificado” caso seja detectado um corante sem correlação com os corantes testados no método. Deve-se testar, em adição aos corantes descritos na norma, a presença do corante carmoisina (INS 122).
Uma nova curva de calibração deve ser obtida a cada batelada de análises.
¶ Densidade relativa a 15°C
¶ Princípio
O tubo oscilante em forma de “U” é uma técnica para determinação da densidade de líquidos e gases baseada na medida eletrônica da frequência de oscilação, a partir da qual o valor da densidade é calculado. O princípio da medida baseia-se no modelo Massa-mola de um oscilador.
¶ Campo de aplicação
Aplica-se a leite fluido.
¶ Materiais e equipamentos
- Densímetro digital de acordo com a norma ISO 15212-1: Oscillation-type density meters - Part 1: Laboratory instruments.
¶ Reagentes e soluções
Não aplicável.
¶ Preparo da amostra
Com o auxílio de um banho-maria, aquecer a amostra a (38 ± 2)°C, mexendo gentilmente até sua homogenização. Analisar ao atingir a temperatura ambiente.
¶ Procedimento de análise
Obter a densidade da amostra a 15°C em g·cm⁻³ de acordo com as instruções do densímetro utilizado.
¶ Expressão dos resultados
Calcular a densidade relativa a 15°C/15°C dividindo-se o valor obtido para a densidade da amostra por 0,99909 g·cm⁻³. Reportar o valor obtido com 3 casas decimais.
¶ Bibliografia
International Organization for Standardization. 15212-1:1998 – Oscillation-type density meters - Part 1: Laboratory instruments. Genebra: 1998.
M. Tanaka, G. Girard, R. Davis, A. Peuto e N. Bignell. Recommended table for the density of water between 0°C and 40°C based on recent experimental reports. Metrologia, v. 38(4), p. 301 – 309, 2001.
¶ Detecção de β-lactoglobulina em queijos frescos
¶ Princípio
O método se baseia na detecção da proteína β-lactoglobulina (BLG), uma das principais proteínas do soro de leite, que é utilizada como marcador de adulteração de queijos frescos produzidos com leite bovino ou bubalino.
A proteína é extraída da matriz, submetida à digestão enzimática com tripsina e convertida em peptídeos característicos. A digestão tríptica da BLG gera o peptídeo marcador TPEVDDEALEK, cuja separação é realizada por cromatografia líquida em fase reversa utilizando coluna C18. A detecção é realizada por espectrometria de massas em modo tandem (LC-MS/MS).
A interpretação do resultado é realizada por comparação da área do pico cromatográfico do peptídeo marcador na amostra com a área obtida em amostra fortificada com BLG no nível de ação estabelecido.
¶ Campo de aplicação
Queijos frescos produzidos com leite bovino e/ou bubalino, exceto produtos ultrafiltrados.
¶ Materiais e equipamentos
- Agitador de tubos tipo vortex;
- Balança analítica;
- Balões volumétricos de 50 mL, 100 mL e 500 mL;
- Centrífuga refrigerada para tubos de 50 mL;
- Coluna para cromatografia líquida com fase estacionária C18 (50 mm × 2,1 mm, partículas de aproximadamente 3,5 μm) ou similar, com pré-coluna C18;
- Estufa com controle de temperatura;
- Mesa agitadora horizontal;
- Micropipetadores e ponteiras para volumes de 10 μL a 10000 μL;
- Microtubos de polipropileno com capacidade de 2 mL;
- Provetas de 100 mL e 500 mL;
- Sistema de ultrassom;
- Sistema de cromatografia líquida acoplada a espectrômetro de massas em modo tandem com fonte de ionização por electrospray (LC-ESI-MS/MS);
- Tubos de polipropileno de 50 mL (tipo falcon);
- Vials de borossilicato para LC.
¶ Reagentes e soluções
- Acetona (C₃H₆O, CAS 67-64-1), mantida a −20°C;
- Acetonitrila (CH₃CN, CAS 75-05-8);
- Ácido fórmico (HCOOH, CAS 64-18-6), pureza mínima de 98%;
- Bicarbonato de amônio (NH₄HCO₃, CAS 1066-33-7) - AMBIC, pureza mínima de 99%;
- Ureia (CH₄N₂O, CAS 57-13-6), pureza mínima de 99,5%;
- Tripsina liofilizada obtida de pâncreas suíno (1000–2000 BAEE units·mg⁻¹);
- β-lactoglobulina bovina, pureza mínima de 90%.
- Fase móvel A: água contendo 0,1% de ácido fórmico;
- Fase móvel B: acetonitrila contendo 0,1% de ácido fórmico;
- Solução de diluição: água e acetonitrila (9:1 v/v) contendo 0,1% de ácido fórmico.
- Solução de ureia 1 mol·L⁻¹;
- Solução de bicarbonato de amônio 50 mmol·L⁻¹;
- Solução tampão de extração (ureia 0,5 mol·L⁻¹; AMBIC 25 mmol·L⁻¹): misturar volumes iguais das soluções de ureia 0,5 mol·L⁻¹ e de AMBIC 25 mmol·L⁻¹L;
- Solução de tripsina 10 g·L⁻¹ (em solução tampão de extração);
- Solução-estoque de β-lactoglobulina (BLG) 10 g·L⁻¹ (em solução tampão de extração);
- Solução de fortificação de β-lactoglobulina digerida (8 g·L⁻¹): em um microtubo de polipropileno de 2 mL, dispensar 500 µL de solução-estoque de BLG 10 g·L⁻¹, adicionar 100 µL de solução de tripsina 10 g·L⁻¹, incubar a 37°C ± 1°C por 16 h e interromper a digestão com 10 µL de ácido fórmico.
Os solventes orgânicos devem ser de grau LC-MS e a água utilizada deve ser de grau ultrapuro.
¶ Preparo da amostra
As amostras de queijo devem ser processadas homogeneizando todo o conteúdo da embalagem, incluindo a parte líquida. Para isso, utilizar um processador de alimentos e/ou ralador até a obtenção de um conteúdo homogêneo.
¶ Procedimento de análise
A - Amostras
- Utilizando balança analítica, pesar aproximadamente 0,5 g de amostra de queijo em tubo de polipropileno de capacidade 50 mL;
- Adicionar 5 mL de acetona gelada (aproximadamente −20°C);
- Agitar em mesa agitadora por cerca de 20 min;
- Centrifugar a 3600 g por 10 min a 4ºC;
- Descartar o sobrenadante e evaporar o resíduo de acetona à temperatura ambiente (aproximadamente 5 min);
- Adicionar 10 mL de solução-tampão de extração e agitar em mesa agitadora por 20 min a 180 rpm;
- Colocar os tubos em banho de ultrassom por 40 min;
- Centrifugar a 3600g por 10 min a 4ºC;
- Adicionar 200 µL de tripsina 10 g·L⁻¹ e homogeneizar em agitador de tubos (~ 10 s);
- Colocar os tubos em estufa a 37°C ± 1°C por 16 h;
- Após a incubação, adicionar 50 µL de ácido fórmico a cada tubo e homogeneizar;
- Transferir alíquotas de 50 µL do sobrenadante para vials e diluir com 950 µL de solução de diluição dos extratos;
- Injetar no sistema LC-MS/MS.
B - Fortificação de queijo com BLG na concentração do nível de ação (350 mg·kg⁻¹) - Controle positivo
- Utilizando balança analítica, pesar aproximadamente 0,5 g de amostra branca de (queijo certificadamente isento de BLG) em tubo de polipropileno de capacidade 50 mL;
- Adicionar 5 mL de acetona gelada (aproximadamente −20°C);
- Agitar em mesa agitadora por cerca de 20 min;
- Centrifugar a 3600g por 10 min a 4ºC;
- Descartar o sobrenadante e evaporar o resíduo de acetona à temperatura ambiente (aproximadamente 5 min);
- Adicionar 35 µL da solução de fortificação de BLG digerida (8 g·L⁻¹);
- Adicionar 10 mL de solução-tampão de extração e agitar em mesa agitadora por 20 min a 180 rpm;
- Colocar os tubos em banho de ultrassom por 40 min;
- Centrifugar a 3600g por 10 min a 4ºC;
- Adicionar 200 µL de tripsina 10 g·L⁻¹ e homogeneizar em agitador de tubos (~ 10 s);
- Colocar os tubos em estufa a 37°C ± 1°C por 16 h;
- Após a incubação, adicionar 50 µL de ácido fórmico a cada tubo e homogeneizar;
- Transferir alíquotas de 50 µL do sobrenadante para vials e diluir com 950 µL de solução de diluição dos extratos;
- Injetar no sistema LC-MS/MS.
Para o controle negativo usa-se a mesma amostra branca de queijo sem a adição da etapa 6 (adição da BLG digerida).
C - Parâmetros instrumentais
A separação cromatográfica é realizada em coluna C18 (50 mm x 2,1 mm, 3,5 µm, 100 Å) ou similar e coluna de guarda C18 (4,0 mm x 3,0 mm, 5 µm), utilizando água (fase móvel A) e acetonitrila (fase móvel B), ambas contendo 0,1% de ácido fórmico, com fluxo aproximado de 300 μL·min⁻¹. A detecção é realizada por espectrometria de massas em modo positivo utilizando ionização por electrospray e monitoramento de reações múltiplas (MRM) do peptídeo marcador TPEVDDEALEK, proveniente da digestão tríptica da β-lactoglobulina.
Parâmetros recomendados:
- Gradiente de eluição: 98% A (0–2 min), 60% A (2–6 min), 35% A (6–7 min), 35% A
(7–7,50 min), 0% A (7,50–9 min), 98% A (9-10 min). - Tempo de auto equilíbrio do sistema: 2 min.
- Temperatura da coluna: 40ºC.
- Volume de injeção: 10 μL.
- Válvula: 0,0 min = descarte / 2,5 min = MS / 6,5 min = descarte.
Parâmetros otimizados do espectrômetro de massas (CE = collision energy; CXP = exit cell potential; DP = declustering potential; EP = entrance potential):
- Analito: TPEVDDEALEK (C₅₂H₈₄N₁₂O₂₃); [M+2H]²⁺.
- Íon precursor: m/z 624; transição de quantificação: m/z 573 (DP = 191 V; EP = 10 V; CE = 22 V; CXP = 29 V).
- Transições de confirmação: m/z 920 (DP = 191 V; EP = 10 V; CE = 44 V; CXP = 29 V); m/z 820 (DP = 191 V; EP = 10 V; CE = 48 V; CXP = 29 V).
- Condições da fonte de ionização: voltagem do ion spray = 5500 V; gás de cortina = 20 psi; gás nebulizador (GS1) = 50 psi; gás auxiliar (GS2) = 45 psi; temperatura = 600ºC; gás de colisão = low.
¶ Expressão dos resultados
O resultado é obtido integrando-se de vale-a-vale o pico do peptídeo TPEVDDEALEK nos cromatogramas das amostras e dos controles. A área do pico do analito de uma amostra deve ser comparada à área do pico do analito nos cromatogramas dos controles negativos e positivos, para que os resultados sejam reportados quanto à presença ou ausência da BLG.
O resultado será negativo quando a área do pico do peptídeo TPEVDDEALEK na amostra for menor que a área do pico do peptídeo TPEVDDEALEK na amostra fortificada no nível de ação (350 mg·kg⁻¹).
O resultado será positivo quando a área do pico do peptídeo TPEVDDEALEK na amostra for maior que a área do pico do peptídeo TPEVDDEALEK na amostra fortificada no nível de ação (350 mg·kg⁻¹).
¶ Bibliografia
Luan Valdemiro Alves de Oliveira, Cristian Rafael Kleemann, Luciano Molognoni, Heitor Daguer, Rodrigo Barcellos Hoff e Elane Schwinden Prudêncio. Reference LC-MS/MS method to detect fresh cheeses adulteration with whey. Food Research International, 156, 111140, 2022.
¶ Detecção de gordura vegetal em produtos lácteos
¶ Princípio
A gordura láctea é saponificada em solução etanólica de hidróxido de potássio e a matéria insaponificável extraída com hexano. Por meio de cromatografia líquida em fase reversa e detecção no ultravioleta, detecta-se a presença/ausência do marcador β-sitosterol.
¶ Campo de aplicação
Este método aplica-se a creme de leite de uso industrial, creme de leite UHT, gordura anidra de leite (butteroil), manteiga, manteiga de garrafa/da terra, ghee, leite em pó integral, leite em pó integral instantâneo e requeijão.
¶ Materiais e equipamentos
-
Balança com resolução mínima de 0,1 g;
-
Banho-maria capaz de manter a temperatura de 80°C, preferencialmente com agitação;
-
Béquer ou erlenmeyer de 50 mL;
-
Centrífuga;
-
Concentrador de amostras, rota-evaporador ou equipamento similar para remoção de solventes orgânicos mediante vácuo;
-
Filtro de membrana de 0,45 μm e seringa;
-
Mesa agitora ou agitador vortex;
-
Micropipeta de 100 μL–1000 μL;
-
Proveta de 25 mL;
-
Sistema cromatográfico, equipado com detector UV a 210 nm e coluna de fase reversa C18 de 250 mm x 4,6 mm e tamanho de partícula de 5 μm;
-
Tubos de centrífuga de 50 mL.
¶ Reagentes e soluções
-
Clorofórmio (CHCl₃, CAS 67-66-3);
-
Fase móvel composta de uma mistura de 5% de 2-propanol ((CH₃)₂CHOH, CAS 67-63-0) em acetonitrila (CH₃CN, CAS 75-05-8), ambos grau HPLC;
-
n-Hexano (n-C₆H₁₄, CAS 110-54-3);
-
Metanol (CH₃OH, CAS 67-56-1);
-
Solução de hidróxido de potássio (KOH, CAS 1310-58-3) a 10% (m/v) em etanol (C₂H₅OH, CAS 64-17-5);
-
Solução de púrpura de bromocresol (C₂₁H₁₆Br₂O₅S, CAS 115-40-2) ou do corante azul patente V (C27H32N2O7S2·Na, CAS 20262-76-4) ou Vermelho Congo (C₃₂H₂₂N₆Na₂O₆S₂, CAS 573-58-0) a 1% ;
-
Solução de referência de β-sitosterol (C₂₉H₅₀O, CAS 83-46-5) a 200 mg·L⁻¹ em etanol. Esta solução tem validade de 2 meses se mantida sob refrigeração;
¶ Preparo da amostra
-
Manteiga, creme de leite cru de uso industrial:
Em tubo de centrífuga de 50 mL, fundir a 50°C quantidade suficiente da amostra para a obtenção de 1 g de gordura. Centrifugar de modo a separar a fase aquosa, obtendo-se uma fase gordurosa isenta de sólidos.
-
Gordura anidra de leite, ghee e manteiga de garrafa/da terra:
Aquecer a amostra em embalagem fechada a aproximadamente 30°C, homogeneizando com agitação manual ou mecânica.
-
Creme de leite UHT, leite em pó integral, leite em pó integral instantâneo e requeijão:
Homogenizar a amostra, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes.
¶ Procedimento de análise
-
Em tubo falcon de 50 mL, pesar:
-
(1,0 ± 0,1) g de gordura para amostras de manteiga e creme de leite cru de uso industrial; ou
-
quantidade de amostra contendo (1,0 ± 0,1) g de gordura.
-
-
Adicionar ao tubo 15 mL de solução etanólica de KOH a 10%, fechando-o em seguida;
-
Levar o tubo ao banho-maria a aproximadamente 80°C por 45 min. Caso o equipamento não possua agitação, agitar as amostras manualmente a cada 5 min;
-
Esfriar, completar o volume para 25 mL com água e, opcionalmente, duas gotas de solução de púrpura de bromocresol ou azul patente V a 1%;
-
Adicionar ao tubo 20 mL de hexano;
-
Agitar por aproximadamente 5 min em mesa agitadora ou agitador vortex;
-
Centrifugar por 5 min a no mínimo 100g para separação das fases;
-
Recolher, com o auxílio de uma pipeta de Pasteur, a fase orgânica superior em um recipiente apropriado ao equipamento utilizado para evaporar o solvente;
-
Evaporar o solvente orgânico sob vácuo, não ultrapassando a temperatura de 40°C;
-
Dissolver a matéria insaponificável obtida em 300 μL de clorofórmio + 500 μL de metanol;
-
Filtrar para vial utilizando seringa e filtro de membrana de 0,45 μm;
-
Injetar a amostra no sistema cromatográfico nas seguintes condições: volume de injeção de 25 μL, fluxo da fase móvel de 1,2 mL·min⁻¹;
-
Em um vial, adicionar 100 μL da solução de referência de β-sitosterol a 200 mg·L⁻¹, 300 μL de clorofórmio e 400 μL de metanol. Injetar no sistema cromatográfico para determinação do tempo de retenção do analito.
-
Se analisando produtos contendo lecitina, adicionar a um vial 250 μL da solução de referência de β-sitosterol a 200 mg·L⁻¹, 300 μL de clorofórmio e 250 μL de metanol e injetar no sistema cromatográfico.
São permitidas alterações no fluxo e tamanho da coluna cromatográfica, desde que mantida a efetiva separação entre o pico do β-sitosterol e demais componentes da matéria insaponificável.
Recomenda-se que o método cromatográfico estenda-se ao menos 10 min após a eluição do β-sitosterol para que ocorra a eluição dos tocoferois, detectáveis apenas com o uso de detector de fluorescência.
¶ Verificação de desempenho
Deve-se comprovar a capacidade de obter-se um limite de detecção de 25 mg·kg⁻¹ de β-sitosterol por meio da análise de 1 g de gordura láctea adicionada de 125 μL da solução de referência de β-sitosterol a 200 mg·L⁻¹.
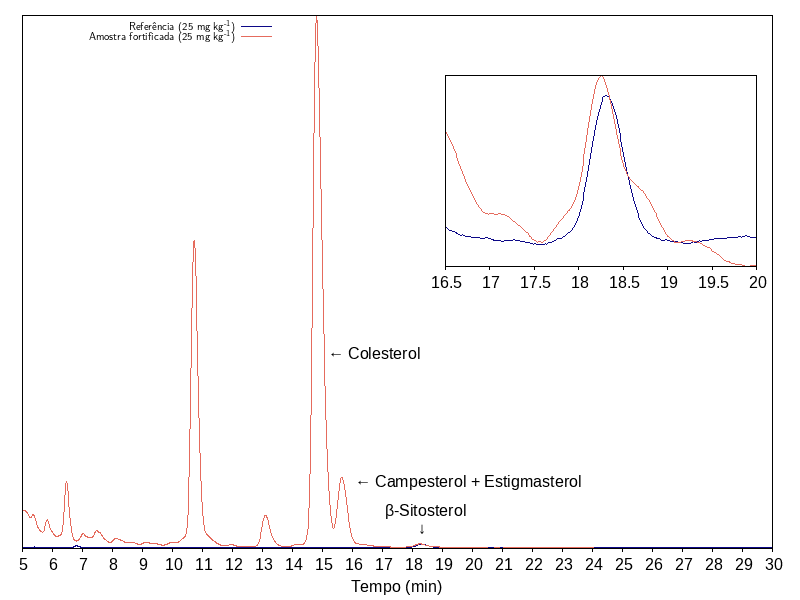
¶ Expressão dos resultados
Integrar o cromatograma da amostra, determinando a altura do pico em relação à linha de base. Reportar resultado “positivo” se o pico cromatográfico na amostra possuir altura superior ao observado na solução de referência de β-sitosterol a 25 mg·L⁻¹ (Procedimento de análise, item 13). Em caso diverso, reportar “negativo”.
Amostras que contenham lecitina serão consideradas “positivas” se a altura do pico em relação à linha de base for superior ao observado na solução de referência de β-sitosterol a 62,5 mg·L⁻¹ (Procedimento de análise, item 14).
¶ Bibliografia
India. Order dated 25th March 2019 related to Method for detection of adulteration in ghee (clarified milk fat) with vegetables oils. Method for Determination of adulteration of Vegetable Oil in Ghee by Reversed Phase High Performance Liquid Chromatography (RP-HPLC). New Delhi: Food Safety and Standards Authority of India, 25 de mar. de 2019. 3 pp.
¶ Detecção de formaldeído
¶ Princípio
O método utiliza o teste de Hehner para a detecção de formaldeído. O teste consiste em uma reação de oxidação do formaldeído e subsequente reação deste com o triptofano da proteína do leite. O desenvolvimento de uma coloração violeta como resultado desta reação indica a presença do aldeído na amostra.
¶ Campo de aplicação
Este método é aplicável a leite fluido.
¶ Materiais e equipamentos
- Micropipeta com capacidade para 100 µL;
- Pipetas volumétricas de 5 mL e 10 mL;
- Tubo de ensaio;
¶ Reagentes e soluções
- Solução de cloreto de férro (III) (FeCl₃, CAS 7705-08-0) a 10% (m/v);
- Solução ácido sulfúrico (H₂SO₄, CAS 7664-93-9) a 80% (v/v);
¶ Preparo da amostra
Homogenizar a amostra a temperatura ambiente, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes. Quando a amostra contiver grumos de creme, aquecer de 38°C a 40°C em banho-maria, esfriar até temperatura ambiente, agitando ocasionalmente.
¶ Procedimento de análise
- Transferir 5 mL da amostra e 5 mL de água para o tubo de ensaio;
- Adicionar 100 µL da solução a 10% de cloreto de ferro (III) e agitar;
- Adicionar, lentamente e pela parede do tubo, 10 mL da solução de ácido sulfúrico a 80%;
- Observar o desenvolvimento de um anel colorido na interface entre o leite e o ácido sulfúrico;
¶ Expressão dos resultados
Reportar “positivo” caso observe-se o desenvolvimento de um anel de coloração violeta.
¶ Bibliografia
Food Safety and Standards Authority of India. Method 01.016 – Detection of formaldehyde in milk. Nova Deli: 2022.
¶ Detecção de peróxido de hidrogênio
¶ Princípio
A peroxidase, ao hidrolisar o peróxido de hidrogênio, libera oxigênio, o qual transformará o guaiacol da sua forma leuco para a forma corada.
¶ Campo de aplicação
Este método é aplicável a leite fluido.
¶ Materiais e equipamentos
-
Banho-maria;
-
Pipetas de 2 mL e 10 mL;
-
Tubo de ensaio.
¶ Reagentes e soluções
-
Leite cru;
-
Solução de peróxido de hidrogênio (H₂O₂, CAS 7722-84-1) a 3% (v/v);
-
Solução hidroalcoólica de guaiacol (C₇H₈O₂, CAS 90-05-1) a 1% (v/v):
Em béquer de 50 mL, colocar 1 mL de guaiacol, adicionar 10 mL de etanol (C₂H₅OH, CAS 64-17-5) 95% e agitar para dissolver. Transferir para balão volumétrico de 100 mL e completar o volume com água. Guardar em frasco âmbar.
¶ Preparo da amostra
Homogenizar a amostra a temperatura ambiente, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes. Quando a amostra contiver grumos de creme, aquecer de 38°C a 40°C em banho-maria, esfriar até temperatura ambiente, agitando ocasionalmente.
¶ Procedimento de análise
-
Transferir 10 mL da amostra e 2 mL de leite cru para um tubo de ensaio, aquecer em banho-maria a 35°C por 5 min, para ativação da enzima peroxidase;
-
Acrescentar 2 mL da solução hidroalcoólica de guaiacol a 1% ao tubo de ensaio, pelas suas paredes;
-
Agitar, aguardar 5 min e observar desenvolvimento de coloração salmão no tubo de ensaio;
-
Efetuar teste “positivo” para verificação dos reagentes adicionando 5 gotas de solução de peróxido de hidrogênio a 3% (v/v) ao tubo. Caso não ocorra a formação de coloração salmão, repetir o teste utilizando outro leite cru como fonte de peroxidase ou nova solução de guaiacol.
¶ Expressão dos resultados
Reportar “positivo” caso observe-se o desenvolvimento de coloração salmão.
¶ Bibliografia
BRASIL. Ministério da Agricultura. Secretaria Nacional de Defesa Agropecuária. Laboratório Nacional de Referência Animal. Métodos analíticos oficiais para controle de produtos de origem animal e seus ingredientes: II – Métodos físicos e químicos. Brasília: 1981.
¶ Detecção de sacarose em leite
¶ Princípio
Fundamenta-se na reação da sacarose com a sacarose-fosforilase. A fosfoglicomutase converte a glicose-1-fostato em glicose-6-fosfato, que é oxidada pela NAD a gliconato-6-fosfato na presença de glicose-6-fosfato-desidrogenase. O NADH formado reduz um sal de tetrazólio a uma formazan de coloração azul, que se determina reflectometricamente.
¶ Campo de aplicação
Este método é aplicável a amostras de leite fluido e leite em pó.
¶ Materiais e equipamentos
-
Balança semi-analítica com resolução mínima de 0,1 g;
-
Equipamento automatizado para medidas reflectométricas (Reflectoquant Merck ou equivalente);
-
Outros materiais necessários para executar o procedimento descrito nas instruções específicas do fabricante do equipamento utilizado.
¶ Reagentes e soluções
-
Reagentes específicos para análise de sacarose em leite no equipamento utilizado, de acordo com as especificações do fabricante. Os reagentes utilizados devem apresentar limite de detecção para sacarose em leite fluido não superior a 0,025% (m/v) e não devem apresentar interferência pela presença de glicose, frutose, citratos e fosfatos para a análise de leite fluido. O limite de detecção e a seletividade dos reagentes utilizados devem ser avaliados e registrados pelo laboratório;
-
Sacarose de pureza conhecida;
-
Matriz branca de leite fluido ou em pó.
¶ Preparo da amostra
-
Preparo de amostras congeladas:
Descongelar a amostra de leite até a temperatura ambiente colocando-a em refrigerador no dia anterior da análise ou utilizando banho-maria entre 30°C e 32°C. Homogenizar a amostra, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes. Quando a amostra contiver grumos de creme, aquecer de 38°C a 40°C em banho-maria, esfriar até temperatura ambiente, agitando ocasionalmente.
-
Preparo de amostras a partir de leite em pó:
Pesar 10,0 g de leite em pó desnatado ou 13,0 g de leite em pó integral, acrescentar 100 mL de água com auxilio de uma pipeta volumétrica e agitar até a completa dissolução. Para leite parcialmente desnatado ou semidesnatado, verificar a forma de reconstituição de acordo com o fabricante.
¶ Procedimento
Calibrar o equipamento conforme orientação do fabricante. Se for necessário clarificar a amostra antes da leitura da reflectância, deve-se garantir que o limite de detecção não seja alterado, isto é, mesmo que seja necessário o procedimento de clarificação da amostra, os reagentes utilizados devem ser capazes de detectar a presença de sacarose em leite fluido com concentração de 0,025% (m/v).
Proceder à execução do procedimento de análise conforme as instruções do fabricante.
A cada batelada de amostras, realizar a análise de uma amostra branca, cujo resultado deve ser negativo, e de uma amostra fortificada com sacarose na concentração de 0,025% (m/v), cujo resultado deve ser positivo.
¶ Expressão dos resultados
Se o resultado direto da medição no equipamento for menor que o limite de detecção, expressar o resultado como “Não detectado”. Se o resultado direto da medição for maior que o limite de detecção, expressar o resultado como “Detectado”.
¶ Bibliografia
Manual do kit Sucrose (Saccharose) – Merck 116141
¶ Determinação de conservantes por cromatografia líquida acoplada à espectrometria de massas
¶ Princípio
O método se baseia na extração sólido-líquido com partição em baixa temperatura, utilizando mistura de solventes orgânicos com baixa proporção de água e acidificados com ácido acético. A separação dos analitos é realizada através da cromatografia líquida em fase reversa utilizando fase estacionária cianopropil. A quantificação é realizada por espectrometria de massas, utilizando fonte de ionização por electrospray. O escopo do método abrange os analitos: ácido benzoico (BEN), ácido sórbico (SOR), natamicina (NAT) e nisina (NIS).
¶ Campo de aplicação
Produtos lácteos (bebidas lácteas, creme de leite, doce de leite e similares, leite condensado, leites fermentados, manteiga, margarina, nata, queijos, ricota, requeijão e sobremesas lácteas).
¶ Materiais e equipamentos
-
Agitador de tubos tipo vortex;
-
Agitador orbital;
-
Balança analítica;
-
Balões volumétricos de 10 mL;
-
Banho de ultrassom;
-
Centrífuga refrigerada para tubos de 15 mL e 50 mL;
-
Centrífuga refrigerada para microtubos;
-
Coluna para cromatografia líquida com fase estacionária di-isopropril-3-cianopropil silano ligado à sílica hidroxilada, com partículas de diâmetro nominal de no mínimo 3,5 μm (Zorbax-Agilent SB-CN, ou similar);
-
Dispensador automático (volume mínimo 10 mL);
-
Homogeneizador/disruptor de amostras (Ultra Turrax ou similar);
-
Micropipetadores e ponteiras para volumes de 2-20 μL; 10-100 μL; 100-1000 μL e 5000-10000 μL;
-
Microtubos de polipropileno, com capacidade para 1,5 mL;
-
Provetas de 250 mL e 500 mL;
-
Sistema de cromatografia líquida acoplada a espectrômetro de massas em modo tandem, com fonte de ionização por electrospray (LC-ESI-MS/MS);
-
Tubos de polipropileno, com capacidade para 15 mL e 50 mL (tipo falcon);
-
Vials de borossilicato para HPLC com capacidade de 1,5 mL.
¶ Reagentes e soluções
-
Solução extratora de metanol (CH₃OH, CAS 67-56-1):água (9:1 v/v) acidificada com 0,1% ácido acético (CH3COOH, CAS 64-19-7).
-
Fase móvel A (solução aquosa de ácido fórmico (HCOOH, CAS 64-18-6) 0,1%).
-
Fase móvel B (solução de acetonitrila (CH₃CN, CAS 75-05-8) acidificada com 0,1% de ácido fórmico).
-
Fase móvel de diluição (fase móvel A:fase móvel B 95:5 v/v).
-
Soluções-estoque de conservantes: nisina A (NIS,C₁₄₃H₂₃₀N₄₂O₃₇S₇, CAS 141-45-5) a 1 g·L⁻¹, preparada em água:acetonitrila (50:50 v/v), acidificada com ácido fórmico em 0,1%; ácido benzoico (BEN, C₆H₅COOH, CAS 65-85-0) a 10 g·L⁻¹, preparada em metanol com baixa proporção de água para dissolução; ácido sórbico (SOR, C₅H₇COOH, CAS 110-44-1) a 1 g·L⁻¹ e 10 g·L⁻¹, natamicina (NAT, C₃₃H₄₇NO₁₃, CAS 7681-93-8) a 100 mg·L⁻¹ e 1 g·L⁻¹, todos preparados em metanol.
-
n-Hexano (n-C₆H₁₄, CAS 110-54-3).
Os solventes orgânicos devem ser de grau LC-MS e a água deve ser de grau ultrapuro. Utilizar banho de ultrassom para dissolução dos padrões.
¶ Preparo da amostra
As amostras de produtos nos quais é autorizado o uso da natamicina devem ser preparadas em duas porções, uma contendo somente a superfície do produto (não ultrapassar 2 mm) e outra contendo a porção interna. As demais amostras devem ser processadas homogeneizando todo o conteúdo. Para isso, utilizar um processador de alimentos e/ou ralador até a obtenção de um conteúdo homogêneo.
¶ Procedimento de análise
¶ Queijos, ricota, requeijão, queijo fundido, bebidas lácteas, leites fermentados, creme de leite e nata
-
Utilizando balança analítica, pesar (2,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL.
-
Adicionar 10 mL de solução de extração aos tubos com amostra.
-
Homogeneizar o conteúdo de cada tubo por volta de cinco segundos no homogeneizador/disruptor.
-
Agitar em mesa agitadora orbital durante 20 min.
-
Centrifugar por 10 min à rotação mínima de 3000 g, sob refrigeração.
-
Transferir o sobrenadante para um tubo de polipropileno de 15 mL de capacidade e levar ao freezer (à temperatura inferior a −18°C) por no mínimo 1 h.
-
Centrifugar novamente por 10 min à rotação mínima de 3000 g, sob refrigeração.
-
Transferir uma alíquota (μl) do sobrenadante do extrato para microtubos de polipropileno de capacidade de 1,5 mL, aplicando-se um fator de diluição de acordo com a sensibilidade do sistema LC-ESI-MS/MS com fase móvel de diluição. Homogeneizar e centrifugar a 13.000 g, sob refrigeração. Por fim, transferir o sobrenadante límpido para vials e injetar no sistema LC-ESI-MS/MS.
-
Preparar uma curva analítica em matriz, pesando-se (2,0 ± 0,1) g “branca” para cada ponto. Estabelecer um gradiente de concentração de acordo com as faixas de trabalho: BEN, 25 mg·kg⁻¹ a 200 mg·kg⁻¹; NAT, 1,25 mg·kg⁻¹ a 10,0 mg·kg⁻¹; SOR, 2,5 mg·kg⁻¹ a 20.0 mg·kg⁻¹ e NIS, 3,13 mg·kg⁻¹ a 25 mg·kg⁻¹. Para isso, adicionar volumes crescentes de 50 μL, 100 μL, 200 μL, 300 μL e 400 μL de cada solução-mix de fortificação e completar o volume com solução de extração q.s.p. 10 mL. Amostras, brancos analíticos, curvas analíticas e demais controles devem estar sujeitos aos mesmos procedimentos analíticos descritos.
¶ Manteiga e margarina
-
Utilizando balança analítica, pesar (2,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL.
-
Adicionar 10 mL de solução de extração aos tubos com amostra.
-
Adicionar 2 mL de n-hexano aos tubos.
-
Agitar cada tubo até dissolver a amostra, utilizando o agitador de tubos (vórtex).
-
Agitar em mesa agitadora orbital durante 20 min.
-
Centrifugar por 10 min à rotação mínima de 3000g, sob refrigeração.
-
Transferir o sobrenadante para um tubo de polipropileno de 15 mL de capacidade e levar ao freezer (à temperatura inferior a −18°C) por no mínimo 1 h.
-
Centrifugar novamente por 10 min à rotação mínima de 3000g, sob refrigeração.
-
Transferir uma alíquota (μl) do sobrenadante do extrato para microtubos de polipropileno de capacidade de 1,5 mL, aplicando-se um fator de diluição de acordo com a sensibilidade do sistema LC-ESI-MS/MS com fase móvel de diluição. Homogeneizar e centrifugar a 13.000 g, sob refrigeração. Por fim, transferir o sobrenadante límpido para vials e injetar no sistema LC-ESI-MS/MS.
-
Preparar uma curva analítica em matriz, pesando-se (2,0 ± 0,1) g de amostra “branca” para cada ponto. Estabelecer um gradiente de concentração de acordo com as faixas de trabalho: BEN, 25 mg·kg⁻¹ a 200 mg·kg⁻¹; NAT, 1,25 mg·kg⁻¹ a 10,0 mg·kg⁻¹; SOR, 2,5 mg·kg⁻¹ a 20.0 mg·kg⁻¹ e NIS, 3,13 mg·kg⁻¹ a 25 mg·kg⁻¹. Para isso, adicionar volumes crescentes de 50 μL, 100 μL, 200 μL, 300 μL e 400 μL de cada solução-mix de fortificação e completar o volume com solução de extração q.s.p. 10 mL. Amostras, brancos analíticos, curvas analíticas e demais controles devem estar sujeitos aos mesmos procedimentos analíticos descritos.
¶ Doce de leite, sobremesas lácteas e leite condensado
-
Utilizando balança analítica, pesar (2,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL.
-
Adicionar 5 mL de solução de extração aos tubos com amostra.
-
Agitar cada tubo até dissolver a amostra, utilizando o agitador de tubos (vórtex).
-
Agitar em mesa agitadora orbital durante 20 min.
-
Centrifugar por 10 min à rotação mínima de 3000 g, sob refrigeração.
-
Transferir o sobrenadante para um tubo de polipropileno de 15 mL de capacidade e levar ao freezer (à temperatura inferior a −18°C) por no mínimo 1 h.
-
Centrifugar novamente por 10 min à rotação mínima de 3000 g, sob refrigeração.
-
Transferir uma alíquota (μl) do sobrenadante do extrato para microtubos de polipropileno de capacidade de 1,5 mL, aplicando-se um fator de diluição de acordo com a sensibilidade do sistema LC-ESI-MS/MS com fase móvel de diluição. Homogeneizar e centrifugar a 13.000 g, sob refrigeração. Por fim, transferir o sobrenadante límpido para vials e injetar no sistema LC-ESI-MS/MS.
-
Preparar uma curva analítica em matriz, pesando-se 2.0 ± 1 g de amostra “branca” para cada ponto. Estabelecer um gradiente de concentração de acordo com as faixas de trabalho: BEN, 25 mg·kg⁻¹ a 200 mg·kg⁻¹; NAT, 1,25 mg·kg⁻¹ a 10,0 mg·kg⁻¹; SOR, 2,5 mg·kg⁻¹ a 20,0 mg·kg⁻¹ e NIS, 3,13 mg·kg⁻¹ a 25 mg·kg⁻¹. Para isso, adicionar volumes crescentes de 30 μL, 60 μL, 120 μL, 180 μL e 240 μL de solução de fortificação SOR 10 g·kg⁻¹ e 25 μL, 50 μL, 100 μL, 150 μL e 200 μL de solução de fortificação NAT 100 mg·kg⁻¹ e completar o volume com solução de extração q.s.p. 10 mL. Amostras, brancos analíticos, curvas analíticas e demais controles devem estar sujeitos aos mesmos procedimentos analíticos descritos.
¶ Parâmetros instrumentais sugeridos
-
Cromatográficos:
-
temperatura do forno de coluna: 35°C;
-
volume de injeção: 10 μL;
-
fluxo da fase móvel: 500 μL min⁻¹;
-
gradiente de eluição: 0 min, 95% A; 2 min, 95% A; 4 min, 15% A, 7 min, 10% A; 8 min, 95% A; com mínimo de 4 min para auto equilíbrio do sistema.
-
| Analitos (sigla) | Íon Precursor (ESI) m/z | Transição de quantificação m/z (EC, V) | Transição de confirmação m/z (EC, V) | DP(V) |
|---|---|---|---|---|
| Ácido sórbico (SOR) | 112,9 (+) | 67,0 (19) | 65,0 (25) | 56 |
| Ácido benzoico (BEN) | 122,9 (+) | 79,0 (17) | 51,0 (51) | 36 |
| Nisina (NIS) | 672,1 (5+) | 811,3 (23) | 649,3 (23) | 56 |
| Natamicina (NAT) | 666,2 (+) | 503,2 (17) | 485,2 (21) | 56 |
¶ Expressão dos resultados
A identificação de cada espécie química é feita pelo tempo de retenção (min) e o perfil de fragmentação molecular, através da razão iônica que deve ser de no mínimo 20% entre as transições (quantificação/confirmação). A quantificação é realizada por relação funcional linear das concentrações (eixo x) dos analitos versus áreas dos picos (eixo y). Expressar o resultado da amostra em “mg/kg” com máximo de quatro algarismos significativos. Para os analitos ácido benzoico e de ácido sórbico, expressar os resultados conforme a Seção Ácido benzoico e/ou benzoatos e Ácido sórbico e/ou sorbatos, respectivamente.
¶ Bibliografia
Luciano Molognoni, Leandro Antunes de Sá Ploêncio, Andressa Camargo Valese, Juliano De Dea Lindner, HeitorDaguer. A simple and fast method for the inspection of preservatives in cheeses and cream by liquid chromatography-electrospray tandem mass spectrometry. Talanta, vol. 147, p. 370-382, 2016.
Luciano Molognoni, Andressa Camargo Valese, Angélica Lorenzetti, Heitor Daguer, Juliano de Dea Lindner. Development of a LC–MS/MS method for the simultaneous determination of sorbic acid, natamycin and tylosin in Dulce de leche. Food Chemistry, vol. 211, p. 741-747, 2018.
Jacson N. Santos, Luciano Molognoni, Thalis A. Vieira, Andressa C. Valese, Patricia Tuzimoto, Cristhiane S. O. Cattani, Heitor Daguer. Scope extension validation of a LC-MS method for the inspection of preservatives in butter. Food Control, vol. 67, p. 209-215, 2016.
¶ Dispersabilidade
Utilizar o método descrito na norma IDF 87, expressando o resultado como um inteiro em “g/100 g”.
¶ Etanol em leites fermentados
Com o auxílio do método descrito na norma AOAC 983.12, determinar o teor de etanol na amostra, expressando o resultado em “% (v/m)” com uma casa decimal.
Poderão ser utilizados equipamentos automatizados de destilação para a execução do método.
¶ Extrato seco desengordurado (ESD), sólidos não gordurosos (SNG)
¶ Leite condensado
Determinar, de acordo com o método apropriado, o teor de gordura e sólidos lácteos totais (Seção Extrato seco total (EST)/Sólidos totais - Leite condensado). Subtrair o teor de gordura do valor de sólidos lácteos totais, ambos expressos na mesma unidade de medida, reportando o valor encontrado com uma casa decimal como "Sólidos lácteos não gordurosos" em “g/100 g”.
¶ Leite fluido e leite em pó
Determinar, de acordo com o método apropriado, o teor de gordura e extrato seco total (EST) na amostra. Subtrair o teor de gordura da amostra do valor de extrato seco total, ambos expressos na mesma unidade de medida, reportando o valor encontrado com uma casa decimal. O extrato seco total de leite em pó é obtido a partir do teor de umidade.
Se necessário converter o teor de gordura de "g/100 mL" para "g/100 g", utilizar a densidade da amostra a 20°C.
¶ Manteiga
Utilizar o método descrito nas normas IDF 80-2 ou IDF 191-2 para obtenção do teor de sólidos não gordurosos (chamado de “teor de insolúveis no éter etílico” para manteiga comum), expressando o resultado obtido em “g/100 g” com duas casas decimais ou uma casa decimal, de acordo com o método utilizado.
Em amostras de manteiga adicionada de cristais de sal, realizar o ensaio em triplicata.
¶ Extrato seco total (EST)/Sólidos totais
¶ Concentrado proteico
Utilizar o método descrito na norma IDF 26 para obtenção da umidade e, por cálculo, do extrato seco total (EST), expressando o resultado obtido em “g/100 g” com duas casas decimais.
¶ Leite e soro de leite líquido
Utilizar o método descrito na norma IDF 21 para obtenção do extrato seco total (EST)/sólidos totais, expressando o resultado obtido em “g/100 g” com duas casas decimais.
¶ Leite condensado
Utilizar o método descrito na norma IDF 15 para obtenção do extrato seco total (EST). Determinar o teor de açúcares de acordo com um dos métodos descritos neste manual. Subtrair do EST o teor de açúcares (exceto lactose) e reportar o resultado obtido como “Sólidos lácteos totais”, expressando o resultado obtido em “g/100 g” com uma casa decimal.
¶ Sobremesa láctea
Utilizar o método descrito na norma IDF 15 para obtenção do extrato seco total (EST), expressando o resultado obtido em “g/100 g” com duas casas decimais.
¶ Fosfatase alcalina
¶ Princípio
A verificação da atividade enzimática é feita mediante a adição à amostra do substrato específico da enzima em condições ideais para sua atuação. A presença do indicador permite identificar a atividade enzimática pela reação colorimétrica com os produtos de degradação.
¶ Campo de aplicação
Este método aplica-se a leite pasteurizado.
¶ Materiais e equipamentos
-
Balança analítica com precisão mínima de 0.001 g;
-
Banho-maria;
-
Pipeta graduada de 5 mL;
-
Tubo de ensaio com tampa.
Obs.: Os tubos de ensaio devem encontrar-se perfeitamente limpos e sem qualquer vestígio de detergentes, em decorrência do processo de lavagem do material.
¶ Reagentes e soluções
-
Catalisador:
Dissolver 0,2 g de sulfato de cobre (II) pentahidratado (CuSO₄·5H₂O, CAS 7758-99-8) em 100 mL de água Tipo I.
-
Solução reagente:
Pesar 0,150 g de 2,6-dicloroquinona-4-cloroimida (C₆H₂Cl₃NO, CAS 101-38-2), dissolver em 50 mL de etanol (C₂H₅OH, CAS 64-17-5), transferir para frasco âmbar e estocar em geladeira. A coloração da solução recentemente preparada é amarelo-citrina, passando a amarelo ouro e tendendo a escurecer, adquirindo tons amarronzados com o envelhecimento. Recomenda-se usar a solução por um período máximo de duas semanas, desde que conservada sob refrigeração e ao abrigo da luz.
-
Substrato:
Pesar 0,5 g de fenilfosfato dissódico dihidratado em um béquer, dissolver com o tampão diluído, transferir para balão volumétrico de 500 mL e completar o volume com o tampão diluído. Recomenda-se usar esta solução durante um período máximo de duas semanas.
-
Tampão carbonato:
Pesar 46,89 g de carbonato de sódio anidro (Na₂CO₃, CAS 497-19-8) e 37,17 g de bicarbonato de sódio (NaHCO₃, CAS 144-55-8), dissolver e levar ao volume de 1000 mL (solução estoque). Retirar uma alíquota de 25 mL da solução estoque, transferir para balão volumétrico de 500 mL e completar o volume. O pH deste tampão diluído deverá situar-se entre 9,5 e 9,7.
¶ Preparo da amostra
Homogenizar a amostra a temperatura ambiente, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes. Quando a amostra contiver grumos de creme, aquecer de 38°C a 40°C em banho-maria, esfriar até temperatura ambiente agitando ocasionalmente. A retirada de uma alíquota da amostra a partir da sua camada superior poderá levar a um resultado positivo ou suspeito, ainda que o leite tenha sido adequadamente pasteurizado. A fosfatase alcalina encontra-se adsorvida aos glóbulos de gordura.
¶ Procedimento de análise
-
Transferir 0,5 mL da amostra a analisar para um tubo de ensaio, adicionar 5 mL do substrato, tampar com rolha de borracha, agitar ligeiramente e levar ao banho-maria mantido entre 39°C e 41°C durante 20 min;
-
Esfriar o tubo de ensaio em água corrente, adicionar 6 gotas de solução reagente e 2 gotas do catalisador;
-
Levar o tubo novamente ao banho-maria entre 39°C e 41°C por 5 min;
-
Repetir o mesmo procedimento acima descrito, usando, em lugar da amostra e em diferentes tubos, 0,5 mL de leite cru e 0,5 mL de leite fervido;
-
Observar a coloração formada.
As provas positivas devem ser repetidas cuidadosamente. A fosfatase alcalina poderá sofrer reativação após algum tempo de pasteurização do leite. Não é comum encontrar esse tipo de interferência, mas o analista deverá ter em conta a vida de prateleira do produto ao ser analisado, principalmente se o teste for conduzido depois de 24 h de o leite ter sido processado.
¶ Expressão dos resultados
Reportar positivo caso observe-se o surgimento de coloração azul intensa no tubo contendo a amostra. O tubo contendo leite cru deve apresentar coloração azul intensa. Reportar negativo caso observe-se uma coloração cinza, equivalente à presente no tubo contendo leite fervido. A tonalidade do azul vai ficando tanto mais intensa, quanto maior for a deficiência de pasteurização.
¶ Bibliografia
BRASIL. Ministério da Agricultura. Secretaria Nacional de Defesa Agropecuária. Laboratório Nacional de Referência Animal. Métodos analíticos oficiais para controle de produtos de origem animal e seus ingredientes: II – Métodos físicos e químicos. Brasília: 1981.
AOAC International. Official Methods of Analysis of AOAC International, Official Method 965.26. 20 ed. Rockville: 2016.
¶ Gordura, matéria gorda, matéria gorda no extrato seco, lipídeos totais
¶ Bebida láctea, creme de leite, doce de leite, leite condensado, leite em pó, leites fermentados e nata
Utilizar o método descrito na norma IDF 249 para obtenção do teor de matéria gorda, expressando o resultado obtido em “g/100 g” com duas casas decimais.
Amostras de creme de leite com elevado teor de emulssificantes, espessantes e estabilizantes devem ser analisadas utilizando-se o médoto IDF 124-3.
¶ Caseínas e caseinatos
Utilizar o método descrito na norma IDF 250 para obtenção do teor de matéria gorda, expressando o resultado obtido em “g/100 g” com duas casas decimais.
¶ Leite fluido
Utilizar o método descrito na norma IDF 249 para obtenção do teor de gordura, expressando o resultado obtido em “g/100 g” com duas casas decimais. Se for necessário expressar o valor em “g/100 mL”, utilizar o valor de densidade a 20°C com três casas decimais para a devida conversão.
Opcionalmente, exceto para leites desnatados, utilizar o método descrito na norma NMKL 40 para obtenção do teor de gordura, expressando o resultado obtido em “g/100 g” com uma casa decimal quando utilizado 10,75 mL de amostra e em “g/100 mL” com uma casa decimal quando utilizado 11,00 mL de amostra.
¶ Manteiga, gordura anidra do leite (butter oil)
Utilizar o método descrito na norma IDF 194 para obtenção do teor de matéria gorda, expressando o resultado obtido em “g/100 g” com duas casas decimais.
Em amostras de manteiga adicionada de cristais de sal, realizar o ensaio em triplicata.
¶ Queijo, requeijão e ricota por coagulação
Utilizar o método descrito na norma IDF 250 ou na norma IDF 222 para obtenção do teor de lipídeos, convertendo o valor obtido em “matéria gorda no extrato seco” a partir da equação:
onde:
L = Teor de lipídios (matéria gorda) na amostra, em g/100 g;
ST = Teor de sólidos totais na amostra obtido por meio da norma IDF 4, em g/100 g.
Expressar o resultado obtido em “g/100 g” com duas casas decimais.
¶ Ricota por concentração
Utilizar o método descrito na norma IDF 249 ou na norma IDF 222 para obtenção do teor de lipídeos, convertendo o valor obtido em “matéria gorda no extrato seco” a partir da equação:
onde:
L = Teor de lipídios (matéria gorda) na amostra, em g/100 g;
ST = Teor de sólidos totais na amostra obtido por meio da norma IDF 58, em g/100 g.
Expressar o resultado obtido em “g/100 g” com duas casas decimais.
¶ Índice crioscópico
Utilizar o método descrito na norma IDF 108 para obtenção do parâmetro depressão do ponto de congelamento, expressando o resultado obtido em “°C” com três casas decimais.
Caso o instrumento utilizado opere apenas em °H, utilizar para conversão entre as unidades a relação: T(°C) = 0,9656 ⋅ T(°H).
¶ Índice de CMP
¶ Princípio
Este método baseia-se na detecção e quantificação de caseínomacropeptídeos (CMP) provenientes da ação proteolítica de enzimas por meio de cromatografia líquida de alta eficiência (CLAE) com separação em coluna de filtração em gel e detecção em ultravioleta (UV).
¶ Campo de aplicação
Leite fluido e leite em pó.
¶ Materiais e equipamentos
-
Agitador magnético;
-
Balança analítica com resolução mínima de 0,0001 g;
-
Balões volumétricos;
-
Banho-maria;
-
Béqueres;
-
Coluna cromatográfica hidrofílica para separação de macromoléculas por filtração em gel com as seguintes características: partículas de sílica esféricas com diâmetro nominal de 4 μm a 4,5 μm, superfície modificada estabilizada com zircônio, diâmetro do poro 150 Å, área superficial 140 m²·g⁻¹, camada hidrofílica mono molecular tipo diol, coluna com 4,6 mm ou 9,4 mm de diâmetro e 250 mm de comprimento;
-
Funil de vidro;
-
Micropipeta;
-
Pipetas volumétricas;
-
Papel de filtro qualitativo;
-
Sistema cromatográfico, equipado com detector UV a 205 nm ou 210 nm, volume de injeção de 20 μL ou 30 μL e com fluxo da fase móvel de 0,5 mL·min⁻¹ a 1,5 mL·min⁻¹.
¶ Reagentes e soluções
-
Solução padrão de caseínomacropeptídeo a 2 g·L⁻¹;
-
Fase móvel tampão fosfato pH 6,0:
Dissolver 1,74 g de hidrogenofosfato de potássio (K2HPO4, CAS 7758-11-4), 12,37 g de dihidrogenofosfato de potássio (KH₂PO₄, CAS 7778-77-0) e 21,41 g de sulfato de sódio (Na₂SO₄, CAS 7757-82-6) em aproximadamente 700 mL de água. Para a utilização de reagentes com molécula de hidratação, fazer as correções nas massas utilizadas. Ajustar o pH da solução para 6,0 usando solução de ácido fosfórico (H₃PO₄, CAS 7664-38-2) a 3 mol·L⁻¹ e solução de hidróxido de potássio (KOH, CAS 1310-58-3) a 3 mol·L⁻¹. Transferir para balão volumétrico de 1 L, completar o volume e filtrar a solução em membrana de 0,45 μm. Antes do uso, desgaseificar.
-
Amostra branca de leite fluido ou leite desidratado. Considera-se, para fins deste método de ensaio, uma amostra branca aquela com Índice de CMP inferior a 5 mg·L⁻¹;
-
Solução de ácido tricloroacético (TCA, CCl₃COOH, CAS 76-03-9) a 24%. Preparar no dia de uso ou a partir de uma solução estoque de ácido tricloroacético a 48%.
¶ Preparo da amostra
-
Preparo de amostras congeladas:
Descongelar a amostra de leite até a temperatura ambiente colocando-a em refrigerador no dia anterior da análise ou utilizando banho-maria entre 30°C e 32°C. Homogenizar a amostra, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes. Quando a amostra contiver grumos de creme, aquecer a 38°C–40°C em banho-maria, esfriar até temperatura ambiente, agitando ocasionalmente.
-
Preparo de amostras de leite em pó:
Pesar 10,0 g de leite em pó desnatado ou 13,0 g de leite em pó integral, acrescentar 100 mL de água com auxilio de uma pipeta volumétrica e agitar até a completa dissolução. Para leite parcialmente desnatado ou semidesnatado, verificar a forma de reconstituição de acordo com o fabricante.
¶ Procedimento de análise
-
Preparar cinco soluções padrão de CMP nas concentrações de 10 mg·L⁻¹, 30 mg·L⁻¹, 40 mg·L⁻¹, 50 mg·L⁻¹, 75 mg·L⁻¹ e 100 mg·L⁻¹ por adição do padrão de CMP à amostra branca;
-
A uma alíquota de 20 mL das amostras preparadas e de cada solução padrão, adicionar 10 mL de ácido tricloroacético a 24% lentamente e sob agitação constante;
-
Deixar em repouso por 60 min à temperatura ambiente e filtrar em papel qualitativo, descartando as primeiras gotas do filtrado. Caso obtenha-se filtrados turvos, filtrar em unidade filtrante com 0,45 μm antes da injeção;
-
Injetar cada solução no sistema cromatográfico, monitorando a 205 ou 210 nm.
Devido à degradação do CMP em solução a 8% de TCA (0,2%/h a 30°C), deverão ser preparadas curvas de calibração a cada batelada de análises.
¶ Cálculo e expressão dos resultados
Calcular a área do pico cromatográfico de cada padrão e do branco. Utilizando o método dos mínimos quadrados, obter uma curva de calibração usando a concentração de CMP em miligramas por litro versus a área do pico cromatográfico de cada padrão subtraída da área do pico na amostra branca. Aceitar a curva para valores de R maiores que 0,95.
Identificar no cromatograma da amostra o pico com o mesmo tempo de retenção dos padrões de CMP e calcular a concentração do analito em miligramas por litro utilizando a equação da curva de calibração.
Expressar o resultado da amostra em “mg/L” como um número inteiro.
¶ Bibliografia
Kees Olieman e J. W. van den Bedem. A sensitive HPLC method of detecting and estimating rennet whey total solids in skim milk powder. Netherlands Milk and Dairy Journal, vol. 37, p. 27-36, 1983.
Kees Olieman e Jan A. M. van Riel. Detection of rennet whey solids in skim milk powder and buttermilk powder with reversed phase HPLC. Netherlands Milk and Dairy Journal, vol. 43, p. 171-184, 1989.
Tarso da Costa Alvim. Efeito da qualidade do leite na detecção de soro lácteo por cromatografia líquida de alto desempenho - filtração gélica (GF-HPLC). Dissertação (Mestrado) - Universidade Federal de Viçosa. Viçosa: 1992.
¶ Índice de CMP em leite condensado
¶ Princípio
Este método baseia-se na detecção e quantificação de caseínomacropeptídeos (CMP) provenientes da ação proteolítica de enzimas por meio de cromatografia líquida de alta eficiência (CLAE) com separação em coluna de filtração em gel e detecção em ultravioleta (UV).
¶ Campo de aplicação
Leite condensado.
¶ Materiais e equipamentos
-
Agitador magnético;
-
Balança analítica com resolução mínima de 0,0001 g;
-
Balões volumétricos;
-
Banho-maria;
-
Béqueres;
-
Coluna cromatográfica hidrofílica para separação de macromoléculas por filtração em gel com as seguintes características: partículas de sílica esféricas com diâmetro nominal de 4 μm a 4,5 μm, superfície modificada estabilizada com zircônio, diâmetro do poro 150 Å, área superficial 140 m²·g⁻¹, camada hidrofílica mono molecular tipo diol, coluna com 4,6 mm ou 9,4 mm de diâmetro e 250 mm de comprimento;
-
Funil de vidro;
-
Micropipeta;
-
Pipetas volumétricas;
-
Papel de filtro qualitativo;
-
Sistema cromatográfico, equipado com detector UV a 205 nm ou 210 nm, volume de injeção de 20 μL ou 30 μL e com fluxo da fase móvel de 0,5 a 1,5 mL·min⁻¹.
¶ Reagentes e soluções
-
Solução padrão de Caseínomacropeptídeo a 2 g·L⁻¹;
-
Fase móvel tampão fosfato pH 6,0:
Dissolver 1,74 g de hidrogenofosfato de potássio (K2HPO4, CAS 7758-11-4), 12,37 g de dihidrogenofosfato de potássio (KH₂PO₄, CAS 7778-77-0) e 21,41 g de sulfato de sódio (Na₂SO₄, CAS 7757-82-6) em aproximadamente 700 mL de água. Para a utilização de reagentes com molécula de hidratação, fazer as correções nas massas utilizadas. Ajustar o pH da solução para 6,0 usando solução de ácido fosfórico (H₃PO₄, CAS 7664-38-2) a 3 mol·L⁻¹ e solução de hidróxido de potássio (KOH, CAS 1310-58-3) a 3 mol·L⁻¹. Transferir para balão volumétrico de 1 L, completar o volume e filtrar a solução em membrana de 0,45 μm. Antes do uso, desgaseificar.
-
Solução de ácido tricloroacético (TCA, CCl₃COOH, CAS 76-03-9) a 24%. Preparar no dia de uso ou a partir de uma solução estoque de ácido tricloroacético a 48%.
¶ Preparo da amostra
-
Leite condensado desnatado:
Reconstituir a amostra utilizando uma parte de leite condensado para 1,73 partes de água, em massa.
-
Leite condensado de alto teor de gordura, integral e semidesnatado:
Reconstituir a amostra utilizando uma parte de leite condensado para 1,38 partes de água, em massa.
¶ Procedimento de análise
-
Adicionar a quatro balões volumétricos de 25 mL, 20 mL da amostra e 0 mL, 0,50 mL, 0,75 mL e 1,00 mL da solução padrão de CMP. Completar para o volume com água;
-
Transferir 20 mL das soluções para béquer de 50 mL e adicionar 10 mL de ácido tricloroacético a 24% lentamente e sob agitação constante;
-
Deixar em repouso por 60 min à temperatura ambiente e filtrar em papel qualitativo, descartando as primeiras gotas do filtrado. Caso obtenha-se filtrados turvos, filtrar em unidade filtrante com 0,45 μm antes da injeção;
-
Injetar cada solução no sistema cromatográfico, monitorando a 205 nm ou 210 nm.
¶ Cálculo e expressão dos resultados
Plotar, no eixo y, as áreas do pico referente ao CMP obtidas dos cromatogramas versus o volume do padrão adicionado (eixo x). Calcular a inclinação (m) e intercepção (b) da curva. Determinar a concentração na amostra a partir da equação:
onde:
b = Intercepção da curva;
C = Concentração do padrão de CMP, em mg/L (2000 mg·L⁻¹);
m = inclinação da curva;
V = volume da amostra adicionado, em mL (20 mL).
Expressar o resultado da amostra em “mg/L” como um número inteiro.
¶ Bibliografia
Kees Olieman e J. W. van den Bedem. A sensitive HPLC method of detecting and estimating rennet whey total solids in skim milk powder. Netherlands Milk and Dairy Journal, vol. 37, p. 27-36, 1983.
Kees Olieman e Jan A. M. van Riel. Detection of rennet whey solids in skim milk powder and buttermilk powder with reversed phase HPLC. Netherlands Milk and Dairy Journal, vol. 43, p. 171-184, 1989.
¶ Índice de insolubilidade
Utilizando o método descrito na norma IDF 129, determinar o Índice de Insolubilidade utilizando temperatura de reconstituição de 24°C. Expressar o resultado com uma casa decimal em “mL (24°C)”.
¶ Índice de peróxidos
¶ Gordura anidra de leite (butter oil)
Utilizar o método descrito na norma IDF 74 para determinação do Índice de Peróxidos em “mmol de O₂/kg de gordura” na amostra, convertendo-o em “meq de O₂/kg de gordura” por meio da seguinte relação: 1 meq = 0,5 mmol. Expressar o resultado final com uma casa decimal.
¶ Manteiga
Adicionar aproximadamente 20 g de amostra a um tubo de centrífuga, fundindo-a em forno estufa entre 40°C e 45°C. Separar a porção gordurosa por centrifugação e filtrar a 40°C–45°C em forno estufa. O filtrado deve apresentar-se límpido e visivelmente livre de umidade e sólidos. Analisar imediatamente de acordo com a metodologia descrita na norma AOAC 965.33, expressando o resultado final em “meq de O₂/kg de gordura” com uma casa decimal.
¶ Lactose
¶ Caseinatos
Determinar o teor de lactose anidra de acordo com o método descrito na norma IDF 106, expressando o resultado obtido em “g/100 g” com duas casas decimais.
¶ Leite fluido, leite em pó e creme de leite
Determinar o teor de lactose anidra de acordo com o método descrito na norma IDF 198, no método descrito na Seção Lactose e sacarose por cromatografia líquida com detecção por índice de refração ou pelo método descrito na Seção Lactose, glicose, frutose e sacarose por cromatografia iônica, expressando o resultado obtido, com duas casas decimais, em “g/100 g”.
¶ Queijo em pó e soro de leite em pó
Determinar o teor de lactose de acordo com o método descrito na Seção Lactose e sacarose por cromatografia líquida com detecção por índice de refração ou pelo método descrito na Seção Lactose, glicose, frutose e sacarose por cromatografia iônica. Reportar os teores de lactose monoidratada em “g/100 g” com duas casas decimais para queijo em pó e lactose em “g/100 g” com duas casas decimais para soro de leite em pó.
¶ Outros produtos lácteos
Determinar o teor de lactose anidra de acordo com o método descrito na Seção Lactose, glicose, frutose e sacarose por cromatografia iônica, expressando o resultado obtido, com duas casas decimais, em “g/100 g”.
¶ Lactose e sacarose por cromatografia líquida com detecção por índice de refração
¶ Princípio
Os carboidratos são dissolvidos em água, precipitando-se as proteínas e gorduras por meio da ação da solução de Carrez, seguido da diluição a um volume apropriado. Após centrifugação, a amostra é novamente diluída com solvente orgânico, filtrada e injetada no cromatógrafo, onde a separação dos açúcares é realizada em modo isocrático em uma coluna de fase estacionária aminopropil silano, utilizando-se um detector de índice de refração para a quantificação.
¶ Campo de aplicação
Este método aplica-se à quantificação dos teores de sacarose e lactose em leite condensado; lactose em leite, leite em pó, queijo em pó e soro de leite em pó; lactose em leite e leite em pó “zero lactose”; e sacarose em leite e leite em pó.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,001 g;
-
Balões volumétricos de 5 mL e 100 mL;
-
Banho de ultrassom;
-
Centrífuga;
-
Coluna para separação de carboidratos com fase estacionária de aminopropil silano, 250 mm × 4,6 mm, tamanho de partícula de 5 μm capaz de separar os picos de interesse;
-
Cromatógrafo líquido equipado com detector por índice de refração capaz de manter fluxo isocrático entre 1,0 mL·min⁻¹ e 1,5 mL·min⁻¹ e injeção de 20 μL;
-
Filtro para seringa de 0,45 μL;
-
Micropipetas de 100 μL e 5000 μL;
-
Pipeta volumétrica de 50 mL;
-
Tubos de centrífuga de 15 mL ou 50 mL.
¶ Reagentes e soluções
-
Fase móvel: solução de acetonitrila (CH₃CN, CAS 75-05-8) : água 75:25;
-
Solução de Carrez I:
Dissolver 10,6 g de hexacianoferrato(II) de potássio (K₄[Fe(CN)₆]·3H₂O, CAS 14459-95-1) em água, transferir para um balão volumétrico de 100 mL e completar para o volume.
-
Solução de Carrez II:
A um balão volumétrico de 100 mL, adicionar 21,9 g de acetato de zinco dihidratado (CH₃COO)₂Zn·2H₂O, CAS 5970-45-6) e 3,2 mL de ácido acético (CH₃COOH, CAS 64-19-7). Diluir para o volume com água.
-
Solução estoque de lactose a 12,5 g·L⁻¹ e sacarose a 7,5 g·L⁻¹ em acetonitrila:água:
Secar os açúcares a 70°C por 4 h em estufa ou de acordo com as instruções do fabricante, se pertinente. A um balão volumétrico de 100 mL, adicionar 1,316 g de lactose monoidratada (C₁₂H₂₂O₁₁·H₂O, CAS 10039-26-6), 0,750 g de sacarose (C₁₂H₂₂O₁₁, CAS 57-50-1) e 50 mL de água. Agitar até dissolução e completar para o volume com acetonitrila.
¶ Preparo da amostra
Homogeneizar a amostra de modo a obter-se alíquota representativa para análise. Amostras de leite em pó devem ser reconstituídas: adicionar a 13,0 g (leite integral) ou 10,0 g (leite desnatado), 100,0 mL de água, agitando até completa dissolução.
¶ Procedimento
-
Curva de calibração, preparada a cada batelada de análises:
-
Para detecção de lactose em leite “zero lactose” ou detecção de sacarose em leite em pó, adicionar 100 μL da solução estoque de lactose e sacarose a balão volumétrico de 5 mL, completando para o volume com solução acetonitrila:água 1:1.
-
Para a quantificação de lactose e sacarose nos demais produtos, adicionar alíquotas de 1 mL, 2 mL, 3 mL e 4 mL da solução estoque de lactose e sacarose a balões volumétricos de 5 mL, completando para o volume com solução acetonitrila:água 1:1. O quinto ponto de calibração é a solução estoque.
-
-
Adicionar a um balão volumétrico de 100 mL:
-
(50,0 ± 0,1) g de leite fluido ou leite em pó reconstituído; ou
-
(2,0 ± 0,1) g de soro de leite em pó e 50 mL de água; ou
-
(10,0 ± 0,1) g de queijo em pó e 50 mL de água; ou
-
(2,0 ± 0,1) g de leite condensado e 50 mL de água.
-
Se necessário, dissolver a amostra por meio de agitação. Amostras de queijo em pó devem ser deixadas em banho de ultrassom por 15 min;
-
Adicionar ao balão volumétrico 2 mL da solução de Carrez I e agitar;
-
Adicionar ao balão volumétrico 2 mL da solução de Carrez II e agitar, completando o volume com água e agitando novamente;
-
Após 5 min, transferir uma parte da solução para tubo de centrífuga;
-
Centrifugar em velocidade suficiente para a separação do precipitado;
-
Tomar uma alíquota de 1 mL do sobrenadante e diluir com igual volume de acetonitrila;
-
Filtrar utilizando filtro de 0,45 μL para vial e injetar 20 μL no sistema cromatográfico em conjunto com a curva de calibração adequada.
As condições cromatográficas poderão ser ajustadas para obter-se melhor separação dos picos, sugerindo-se utilizar uma fase móvel acetonitrila:água entre 75:25 e 85:15, fluxo entre 1,0 mL min⁻¹ e 1,5 mL min⁻¹, temperatura do detector e coluna em 35°C. O detector deve ser ajustado de maneira a obter-se uma relação sinal/ruído superior a 3 para o menor pico da curva de calibração. A Figura 2 demonstra a separação entre diversos açúcares.
¶ Expressão dos resultados
Para detecção de lactose em leite “zero lactose” ou detecção de sacarose em leite em pó, obter a concentração de sacarose e/ou lactose na amostra utilizando a equação:
onde:
Aam = área do pico da amostra no cromatograma;
Ap = área do pico do padrão no cromatograma;
m = massa da amostra, em g;
Vb = volume do balão volumétrico utilizado na diluição inicial, em mL (100 mL);
0,1 = fator de conversão de g/kg para g/100 g;
0,02 = concentração do padrão, em g·L⁻¹;
2 = fator de diluição.
Para a quantificação de lactose e sacarose nos demais produtos, obter pelo método dos mínimos quadrados a equação da curva de calibração a partir da área dos picos no cromatograma e concentração dos padrões em g l⁻¹. A partir desta equação, obter a concentração de sacarose e/ou lactose na amostra utilizando a equação:
onde:
W = concentração de lactose/sacarose obtida da curva de calibração;
Vb = volume do balão volumétrico utilizado na diluição inicial, em mL (100 mL);
m = massa da amostra, em g;
2 = fator de diluição;
0,1 = fator de conversão de g/kg para g/100 g.
Se necessário expressar o resultado em lactose monoidratada, multiplicar o valor obtido por 1,0526.
Expressar o resultado com duas casas decimais.
¶ Bibliografia
Nordic Committee on Food Analysis. Method 148 – Fructose, glucose and saccharose. Liquid chromatographic determination in fruit and vegetable products. Espoo: 1993.
International Organization for Standardization. ISO 22184:2021 – Milk and milk products. Determination of the sugar contents. High performance anion exchange chromatography with pulsed amperometric detection method (HPAEC-PAD). Genebra: 2021.
¶ Lactose, glicose, frutose e sacarose por cromatografia iônica
¶ Princípio
Os contra-íons de aminas interagem com a espécie aniônica, de forma que quanto maior o pKa da espécie, maior o tempo de retenção do composto na coluna, pois é favorecida a interação entre o analito e a fase estacionária. Existe uma hierarquia de acidez que preconiza o aumento da interação com a fase estacionária, baseada na relação do pKa e da elevação do pH, por interação com a fase móvel. Quanto maior o número de hidroxilas (-OH), carbonilas (C=O) ou heteroátomos de oxigênio (R-O-R) presentes nas cadeias de carboidratos, maior será a interação com a fase estacionária, aumentando o tempo de retenção e permitindo a separação dos sacarídeos.
¶ Campo de aplicação
Este método aplica-se a leite, produtos lácteos e mel.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,01 g;
-
Balões volumétricos de 25 mL, 50 mL,100 mL, 1 L, 2 L;
-
Duas colunas para carboidratos de estireno/divinilbenzeno montadas em série;
-
Cromatógrafo de íons, com detector amperométrico de célula de ouro, com sistema de diálise;
-
Micropipetas de 100 μL, 1000 μL e 5000 μL;
-
Tubos para cromatografia iônica.
¶ Reagentes e soluções
-
Fase móvel: solução de hidróxido de sódio (NaOH, CAS 1310-73-2) a 300 mmol·L⁻¹ e acetato de sódio (CH₃COONa, CAS 127-09-3) a 1 mmol·L⁻¹;
-
Padrão de frutose a 2 g·L⁻¹;
-
Padrão de glicose a 2 g·L⁻¹;
-
Padrão de lactose a 2 g·L⁻¹;
-
Padrão de sacarose a 2 g·L⁻¹.
¶ Preparo da amostra
-
Lactose em leite fluido:
Pesar uma alíquota de 0,100 g, transferir para balão volumétrico de 100 mL e completar para o volume. Preparar a amostra em duplicata. Para produtos isentos ou com teor reduzido de lactose, utilizar uma alíquota de 1,00 g.
-
Sacarose em leite em pó:
Pesar 10,0 g de leite em pó desnatado ou 13,0 g de leite em pó integral, acrescentar 100 mL de água e agitar até a completa dissolução. Para leite parcialmente desnatado ou semidesnatado, verificar a forma de reconstituição de acordo com o fabricante. Aliquotar 1,0 mL, transferir para balão volumétrico de 100 mL e completar para o volume.
-
Sacarose em leite fluido
Aliquotar 1,0 mL, transferir para balão volumétrico de 100 mL e completar para o volume.
-
Lactose, sacarose, glicose e frutose em leite condensado:
Pesar uma alíquota de 0,5 g, transferir para balão volumétrico de 100 mL e completar para o volume. Após, diluir 1,0 mL em balão volumétrico de 25 mL. Preparar a amostra em duplicata.
-
Lactose em leite em pó e soro de leite em pó
Pesar uma alíquota de 0,25 g, transferir para balão volumétrico de 100 mL e completar para o volume. Após, diluir 1,0 mL em balão volumétrico de 25 mL. Preparar a amostra em duplicata
-
Mel
Pesar uma alíquota de 0,50 g, transferir para balão volumétrico de 100 mL e completar para o volume. Após, diluir 1,0 mL em balão volumétrico de 25 mL. Preparar a amostra em duplicata
Pode-se efetuar ajustes nas diluições, de modo que as leituras obtidas estejam dentro da faixa linear da curva de calibração.
¶ Procedimento
-
Equilibrar o sistema cromatográfico nas seguintes condições: fluxo isocrático entre 0,3 mL·min⁻¹ e 0,4 mL·min⁻¹, eletrodo de trabalho de ouro (3 mm), eletrodo de referência de paládio, espaçador em 50 μm, potencial de medida em 50 mV, faixa de medida em 200 μA, duração da medida em 100 ms, duração do ciclo em 550 ms, temperatura da célula de medida em 35°C no modo PAD, loop de injeção de 20 μL;
-
Preparar, em duplicata, uma curva de calibração dos açúcares por diluição das soluções padrão nas concentrações individuais de 5 mg·L⁻¹, 10 mg·L⁻¹, 30 mg·L⁻¹, 50 mg·L⁻¹, 70 mg·L⁻¹ e 90 mg·L⁻¹. O coeficiente de correlação (r²) da curva de calibração deve ser superior a 0,98.
-
Transferir as duplicatas das amostras preparadas para vials e injetar.
A cada 15 amostras, deve-se verificar a validade da curva utilizando-se o ponto central da mesma, tolerando-se uma variação de ± 5% nesta concentração. Caso encontre-se uma variação superior, efetuar nova calibração reinjetando a curva e o controle.
¶ Expressão dos resultados
Os resultados são calculados a partir da equação da reta obtida na curva de calibração, calculando-se a concentração baseada na respectiva resposta.
Produtos isentos ou com baixo teor de lactose:
onde:
c = valor obtido da curva de concentração, em mg l⁻¹;
m = massa da amostra, em g;
v = volume da amostra, em mL;
V = volume do balão volumétrico utilizado no preparo da amostra;
10000 = fator de conversão de mg·kg⁻¹ para g/100 g.
10 = fator de conversão de mg·kg⁻¹ para mg/100 g.
f = fator de diluição adicional, se utilizado.
Para leite e produtos lácteos, expressar o resultado obtido com duas casas decimais. Para sacarose em leite fluido e desidratado, reportar “detectado” se a concentração encontrada for superior a 0,025 g/100 mL. Em caso diverso, reportar “não detectado”.
Para mel, reportar o teor de "açúcares redutores" como a soma dos teores de glicose e frutose em "g/100g" com uma casa decimal. Reportar o teor de sacarose em "g/100g" com uma casa decimal.
¶ Bibliografia
Metrohm. Determinação de Lactose Residual em leite zero lactose. AN-C-BR-002. Disponível em http://www.metrohm.com, acessada em 31/08/2016.
¶ Maltodextrina
¶ Princípio
São precipitados os interferentes e a maltodextrina (HEX5) é determinada por cromatografia líquida acoplada a espectrometria de massas.
¶ Campo de aplicação
Produtos lácteos.
¶ Materiais e equipamentos
-
Agitador de tubos tipo Vortex ou similar;
-
Balança analítica com resolução mínima de 0,0001 g;
-
Balões volumétricos;
-
Béquer de 100 mL;
-
Centrífuga para Eppendorf refrigerada;
-
Centrífuga refrigerada;
-
Coluna de HPLC HILIC, 150 x 2,1 mm, tamanho de partícula de 5 μm;
-
Frasco Autoclavável de 250 mL;
-
Homogeneizador tipo turrax;
-
Micropipetas de 0,5 μL a 5 μL, 10 μL a 100 μL e 100 μL a 1000 μL;
-
Proveta 50 mL;
-
Sistema de cromatografia líquida com detecção de massas MS/MS triplo quadrupolo;
-
Tubos de centrífuga de 15 mL e 50 mL, de polipropileno, com tampa de rosca;
-
Tubos Eppendorf de 1,0 mL a 2.0 mL;
-
Vial com capacidade de 1,5 mL a 2.0 mL;
¶ Reagentes e soluções
-
Acetonitrila (CH₃CN, CAS 75-05-8), grau HPLC;
-
Fase Móvel A:
Adicionar 20 mL da solução de acetato de amônio (CH₃COONH₄, CAS 631-61-8) 100 mmol·L⁻¹ e transferir quantitativamente para um balão volumétrico de 1000 mL e adicionar um pouco de água ultrapura. Adicionar 1 mL de ácido acético (CH3COOH, CAS 64-19-7) glacial ao balão volumétrico e completar com água para o volume.
-
Fase Móvel B:
Adicionar 20 mL da solução de acetato de amônio 100 mmol·L⁻¹ e transferir quantitativamente para um balão volumétrico de 1000 mL e adicionar um pouco de acetonitrila. Adicionar 1 mL de ácido acético glacial ao balão volumétrico e completar com acetonitrila para o volume.
-
Leite fluido isento de maltodextrinas;
-
Maltodextrina dextrose equivalente (DE) 16,6-19,5;
-
Solução de acetato de amônio 100 mmol·L⁻¹, válida por 3 meses sob refrigeração;
-
Solução de ácido tricloroacético (TCA, CCl₃COOH, CAS 76-03-9) 5%. Válida por 1 mês se estocada em refrigerador com temperatura inferior a 10°C;
-
Solução estoque de 100 g l⁻¹ de maltodextrina dextrose equivalente (DE) 16,6-19,5. Válida por 1 mês se estocada em refrigerador com temperatura inferior a 10°C;
¶ Preparo da amostra
-
Leite fluido:
Homogeneizar a amostra.
-
Leite em pó:
Pesar (1,00 ± 0,01) g de leite em pó desnatado ou (1,30 ± 0,01) g de leite em pó integral em tubo falcon de 50 mL. Reconstituir o leite com 10 mL de água. Para leite parcialmente desnatado ou semidesnatado, verificar a forma de reconstituição de acordo com as instruções do fabricante.
-
Queijo e Iogurte:
Pesar, em tubo falcon de 50 mL, (5,00 ± 0,10) g da amostra. Diluir com 10 mL de TCA 5%.
-
Outros produtos lácteos:
Pesar, em em copo de béquer, (5,00 ± 0,10) g da amostra. Diluir com 50 mL de água ultrapura morna (≈ 50°C). Deixar esfriar.
¶ Procedimento de análise
-
Aliquotar 1 mL de amostra para tubos falcon de 15 mL;
-
Adicionar 500 μL de TCA a 5% e agitar manualmente ou com vórtex;
-
Adicionar mais 500 μL de TCA a 5% e agitar;
-
Adicionar 1 mL de TCA a 5% e agitar;
-
Centrifugar a 4000 rpm por 20 min a 5°C;
-
Transferir, com auxílio de micropipeta, 1 mL do sobrenadante para tubo de eppendorf;
-
Centrifugar a 12000 rpm por 20 min a 0°C;
-
Transferir 900 μL do sobrenadante para os vials e analisar por LC-MS/MS. Para queijos e iogurte transferir 500 μL do sobrenadante do Eppendorf para os vials. Completar os vials com 500 μL de TCA a 5%. Agitar os vials e analisar por LC-MS/MS;
-
Analisar, por batelada, uma alíquota de 1 mL de leite fluido fortificado a 0,25 g·L⁻¹ para verificação do limite de detecção.
Ajustar o sistema cromatográfico e detector MS-MS de acordo com as condições sugeridas nas Tabelas 4 e 5. Ajustes nestas condições visando otimizar o sinal do analito podem ser necessários.
| Tempo, em min | A, em % | B, em % |
|---|---|---|
| 0,0 | 5 | 95 |
| 4,0 | 70 | 30 |
| 6,5 | 70 | 30 |
| 8,0 | 5 | 95 |
| 13,0 | 5 | 95 |
| m/z Q1 | m/z Q3 | Dwell Time | DP | EP | CE | CXP |
|---|---|---|---|---|---|---|
| 851,21 | 527,1 | 200 | 296 | 10 | 67,000 | 4,000 |
| 851,21 | 689,1 | 200 | 296 | 10 | 67,000 | 6,000 |
| 851,21 | 509,0 | 200 | 296 | 10 | 67,000 | 4,000 |
| 851,21 | 365,0 | 200 | 296 | 10 | 79,000 | 4,000 |
| 851,21 | 671,0 | 200 | 296 | 10 | 63,000 | 4,000 |
¶ Expressão dos resultados
Se a resposta obtida na amostra for igual ou superior à obtida na amostra fortificada a 0,25 g·L⁻¹, o resultado é considerado positivo para a presença de maltodextrina na amostra. Caso contrário, o resultado é considerado negativo.
¶ Bibliografia
Gustavo Braga Sanvido, Jerusa Simone Garcia, Yuri E. Corilo, Boniek G. Vaz, Jorge J. Zacca, Ricardo G. Cosso, Marcos N. Eberlin, e Martin G. Peter. Fast Screening and Secure Confirmation of Milk Powder Adulteration with Maltodextrin Via Electrospray Ionization-Mass Spectrometry [ESI(+)-MS] and Selective Enzymatic Hydrolysis. Journal of Agricultural and Food Chemistry, vol. 58, p. 9407-9412, 2010.
¶ Natamicina
Seguir o procedimento descrito nas normas IDF 140-1, IDF 140-2 ou o procedimento descrito na Seção Determinação de conservantes por cromatografia líquida acoplada à espectrometria de massas, conforme a matriz a ser analisada. Reportar o resultado obtido na camada exterior em “mg/dm²” e na massa em “mg/kg”.
¶ Nitritos e nitratos em queijos
Determinar o teor de nitritos e nitratos utilizando o método descrito nas normas NMKL 165, NMKL 194 ou IDF 189-1. Reportar o resultado conforme descrito abaixo:
Nitritos
-
Resultados inferiores a 10 mg de NaNO₂/kg devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores a 10 mg de NaNO₂/kg e inferiores a 20 mg de NaNO₂/kg devem ser expressos como “detectado”;
-
Resultados maiores ou iguais a 20 mg de NaNO₂/kg devem ser expressos arredondados para um número inteiro.
Nitratos
-
Resultados inferiores a 10 mg de NaNO₃/kg devem ser expressos como “não detectado”;
-
Resultados iguais ou superiores a 10 mg de NaNO₃/kg e inferiores a 30 mg de /kg devem ser expressos como “detectado”;
-
Resultados maiores ou iguais a 30 mg de NaNO₃/kg devem ser expressos arredondados para um número inteiro.
Uma nova curva de calibração deve ser obtida a cada batelada de análises, independentemente do método utilizado.
¶ Nisina
Seguir o procedimento descrito na norma IDF/RM 217 ou, opcionalmente, o procedimento descrito na Seção Determinação de conservantes por cromatografia líquida acoplada à espectrometria de massas. Reportar o resultado obtido com uma casa decimal em “mg/kg”.
¶ Partículas queimadas
¶ Caseínas e caseinatos
Seguir o procedimento descrito na norma IDF 107, reportando como resultado a classificação observada, ou seja, como “Disco A”, “Disco B”, “Disco C” ou “Disco D”.
¶ Leite em pó
Seguir o procedimento descrito no ADPI Bulletin 916, reportando como resultado a classificação observada, ou seja, como “Disco A”, “Disco B”, “Disco C” ou “Disco D”.
¶ Peroxidase
¶ Princípio
A peroxidase, ao hidrolisar o peróxido de hidrogênio, libera oxigênio, o qual transformará o guaiacol da sua forma leuco para a forma corada.
¶ Campo de aplicação
Este método é aplicável a leite pasteurizado.
¶ Materiais e equipamentos
-
Banho-maria;
-
Pipetas de 2 mL e 10 mL;
-
Tubo de ensaio.
¶ Reagentes e soluções
-
Solução de peróxido de hidrogênio (H₂O₂, CAS 7722-84-1) a 3% (v/v);
-
Solução hidroalcoólica de guaiacol a 1% (v/v):
Em béquer de 50 mL, colocar 1 mL de guaiacol (C₇H₈O₂, CAS 90-05-1), adicionar 10 mL de etanol (C₂H₅OH, CAS 64-17-5) e agitar para dissolver. Transferir para balão volumétrico de 100 mL e completar o volume com água. Guardar em frasco âmbar.
¶ Preparo da amostra
Homogenizar a amostra a temperatura ambiente, agitando e invertendo o recipiente ou embalagem cinco ou seis vezes. Quando a amostra contiver grumos de creme, aquecer de 38°C a 40°C em banho-maria, esfriar até temperatura ambiente, agitando ocasionalmente.
¶ Procedimento de análise
-
Transferir 10 mL da amostra para um tubo de ensaio, aquecer em banho-maria a 45°C por 5 min, para ativação da enzima;
-
Acrescentar 2 mL da solução hidroalcoólica de guaiacol a 1% ao tubo de ensaio, pelas suas paredes;
-
Adicionar três gotas da solução de peróxido de hidrogênio a 3%;
-
Aguardar 5 min e observar desenvolvimento de um anel de coloração salmão no tubo de ensaio.
¶ Expressão dos resultados
Reportar positivo caso observe-se o desenvolvimento de um anel de coloração salmão.
¶ Bibliografia
BRASIL. Ministério da Agricultura. Secretaria Nacional de Defesa Agropecuária. Laboratório Nacional de Referência Animal. Métodos analíticos oficiais para controle de produtos de origem animal e seus ingredientes: II – Métodos físicos e químicos. Brasília: 1981.
¶ pH
¶ Princípio
Fundamenta-se na medida, por meio de aparelho adequado, da concentração de íons hidrogênio na amostra.
¶ Campo de aplicação
Soro de leite, soro de leite em pó, ovos líquidos, ovos desidratados, gelatina e colágeno.
¶ Materiais e equipamentos
-
Balança com resolução mínima de 0,1 g;
-
Béquer de 100 mL;
-
pH-metro;
-
Proveta.
¶ Reagentes e soluções
- Soluções padrão de pH.
¶ Preparo da amostra
-
Soro de leite em pó, gelatina e colágeno: preparar uma solução a 10% (m/v);
-
Soro de leite: homogeneizar;
-
Ovos líquidos: homogeneizar;
-
Ovos desidratados: reconstituir na proporção em massa de 1 parte do produto para 3 partes de água livre de ;
¶ Procedimento de análise
-
Calibrar o pH-metro de acordo com as instruções do fabricante utilizando um mínimo de dois padrões abrangendo a faixa de pH a ser medida;
-
Efetuar a medida do pH, com correção para 20°C.
¶ Expressão dos resultados
Reportar o valor obtido com uma casa decimal.
¶ Bibliografia
BRASIL. Ministério da Agricultura. Secretaria Nacional de Defesa Agropecuária. Laboratório Nacional de Referência Animal. Métodos analíticos oficiais para controle de produtos de origem animal e seus ingredientes: II – Métodos físicos e químicos. Brasília: 1981.
¶ Proteína
Determinar o teor de proteína bruta na amostra de acordo com a norma ISO 1871 ou IDF 20-1. Expressar o resultado obtido em “g/100 g” com duas casas decimais. Alternativamente, utilizar o método descrito na norma IDF 185. O fator de conversão de nitrogênio para proteína a ser utilizado é de 6,38 em todos os métodos.
Para amostras de leite em pó, expressar o resultado com base no extrato seco desengordurado:
onde:
P = Teor de proteína, em g/100 g;
ESD = Teor de extrato seco desengordurado, obtido por meio dos métodos descritos neste Manual, em g/100 g.
Para amostras de leite condensado, expressar o resultado com base no extrato seco desengordurado de origem láctea:
onde:
P = Teor de proteína, em g/100 g;
ESD = Teor de extrato seco desengordurado de origem láctea, obtido por meio dos métodos descritos neste Manual, em g/100 g.
Para amostras de caseína e concentrados proteicos, expressar o resultado em base seca:
onde:
P = Teor de proteína, em g/100 g;
EST = Teor de extrato seco total, obtido por meio dos métodos descrito neste Manual, em g/100 g.
Para compostos lácteos em que conste o procedimento de reconstituição do produto, o mesmo deverá ser analisado em sua apresentação em pó e após reconstituição, reportando os valores obtidos em “g/100 g” e “g/100 mL”, respectivamente.
¶ Substâncias redutoras voláteis (álcool etílico)
¶ Princípio
Em meio ácido, hidroxilas ligadas a carbono primário ou secundário são oxidados pela ação do ácido crômico, com consequente redução do cromo(VI) a cromo(III), modificando a coloração da solução sulfocrômica.
¶ Campo de aplicação
Este método aplica-se a leite fluido.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,01 g;
-
Bico de Bünsen ou placa aquecedora;
-
Kitazato de 500 mL ou balão de fundo redondo com saída lateral de 500 mL;
-
Pipeta graduada de 10 mL;
-
Pipeta de Pasteur;
-
Proveta de 100 mL;
-
Rolha de borracha, para vedação da abertura superior do kitassato/balão com saída lateral;
-
Tubo de ensaio 20 x 200 mm;
-
Tubo de silicone ou látex de 25 cm.
¶ Reagentes e soluções
-
Antiespumante (solução a 3%);
-
Solução sulfocrômica:
Dissolver 1,15 g de dicromato de potássio (K₂Cr₂O₇, CAS 7778-50-9) em 10 mL de água, transferir para balão volumétrico de 100 mL e completar o volume com ácido sulfúrico concentrado (ácido sulfúrico (H₂SO₄, CAS 7664-93-9)) a 98%.
¶ Preparo da amostra
Homogenizar a amostra a temperatura ambiente, agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes. Quando a amostra contiver grumos de creme, aquecer de 38°C a 40°C em banho-maria, esfriar até temperatura ambiente, agitando ocasionalmente.
¶ Procedimento de análise
-
Medir 100 mL da amostra e transferir para o kitassato ou balão de fundo redondo com saída lateral;
-
Adicionar 10 mL de antiespumante e misturar bem;
-
Transferir para um tubo de ensaio 2 mL da solução sulfocrômica e mergulhar nessa solução a extremidade da pipeta de Pasteur acoplado ao kitassato/balão por um tubo de silicone ou látex, de modo a formar um sistema fechado;
-
Aquecer a amostra contida no kitassato/balão, mantendo em fervura por 5 min;
-
Observar a coloração da solução sulfocrômica no tubo de ensaio.
¶ Expressão dos resultados
Reportar “positivo” se observada a mudança na coloração da solução sulfocrômica para verde e “negativo” em caso diverso.
Dado que o formaldeído é um interferente para este ensaio, não realizá-lo se a amostras obtiver resultado positivo para formaldeído.
¶ Bibliografia
Vladimir N. Alexeyev. Qualitative Analysis, Editora Mir. Moscou: 1967.
¶ Umectabilidade
Utilizar o método descrito na norma IDF 87, expressando o resultado como um inteiro em “s”.
¶ Umidade
¶ Caseína e caseinatos
Utilizar o método descrito na norma IDF 78, expressando o resultado em “g/100 g” com duas casas decimais.
¶ Doce de leite
Utilizar o método descrito na norma IDF 15 para obtenção do teor de sólidos totais na amostra. Calcular o teor de umidade:
onde ST é o teor de sólidos totais. Expressar o resultado obtido em “g/100 g” com duas casas decimais.
¶ Gordura anidra do leite (butter oil)
Utilizar o método descrito na norma IDF 23, obtendo a quantidade de água na amostra. Reportar o valor obtido como Umidade, expressando o resultado em “g/100 g” com duas casas decimais.
¶ Manteiga
Utilizar o método descrito nas normas IDF 80-1 ou IDF 191-1 para obtenção do teor de umidade, expressando o resultado obtido em “g/100 g” com duas casas decimais ou uma casa decimal, de acordo com o método utilizado.
¶ Leite em pó, soro de leite em pó, concentrados proteicos em pó e queijo em pó
Utilizar o método descrito na norma IDF 26, expressando o resultado em “g/100 g” com duas casas decimais.
¶ Queijo, requeijão e ricota por coagulação
Utilizar o método descrito na norma IDF 4 para obtenção do teor de sólidos totais na amostra. Calcular o teor de umidade:
onde ST é o teor de sólidos totais. Expressar o resultado obtido em “g/100 g” com duas casas decimais.
¶ Ricota por concentração
Utilizar o método descrito na norma IDF 58 para obtenção do teor de sólidos totais na amostra. Calcular o teor de umidade:
onde ST é o teor de sólidos totais. Expressar o resultado obtido em “g/100 g” com duas casas decimais.
¶ Umidade e lipídios em leite e produtos lácteos por microondas e ressonância magnética nuclear - método de rotina
Pode-se utilizar, como método de rotina, o método descrito na norma AOAC 2008.06 para determinação dos teores de umidade e gordura no leite e produtos lácteos, à exceção de doce de leite, leite condensado e sobremesas lácteas.
Resultados não conformes devem ser confirmados por outro método descrito neste manual.
¶ Mel e produtos apícolas
¶ Preparo de amostras
Para amostras de mel, devem ser seguidos os procedimento descritos na norma ABNT NBR 15714-1. Demais produtos devem seguir os procedimentos descritos nos métodos.
¶ Acidez livre
Determinar a acidez livre utilizando o método descrito nas normas ABNT NBR 15714-6 ou AOAC 962.19, reportando o resultado obtido em “meq/kg” com uma casa decimal.
¶ Açúcares redutores
Determinar os teores de glicose e frutose pelo método AOAC 977.20 ou pelo método descrito na Seção Lactose, glicose, frutose e sacarose por cromatografia iônica, reportando como “Açúcares redutores” a soma dos valores obtidos em “g/100 g” com uma casa decimal.
¶ Atividade diastásica
Determinar a atividade diastásica utilizando o método descrito na norma AOAC 958.09. Reportar o resultado obtido na escala Goethe com uma casa decimal.
¶ Compostos fenólicos em própolis
Determinar o teor de compostos fenólicos totais utilizando o método descrito na norma ABNT NBR 16956-6, reportando o resultado obtido em “g/100 g” com duas casas decimais.
¶ Compostos fenólicos em extrato de própolis
¶ Princípio
Baseia-se na reação dos compostos fenólicos com o reagente de Folin-Ciocalteau e posterior quantificação espectrofotométrica a 760 nm. O teor de compostos fenólicos totais é expresso em g de ácido gálico/100 g.
¶ Campo de aplicação
Este método é aplicável a extrato de própolis.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,0001 g;
-
Balões volumétricos de 10 mL, 50 mL e 100 mL;
-
Espectrofotômetro de absorção molecular capaz de efetuar leituras em 760 nm;
-
Micropipetador de 100 μL–1000 μL.
¶ Reagentes e soluções
-
Etanol (C₂H₅OH, CAS 64-17-5);
-
Reagente de Folin-Ciocalteau 2 mol·L⁻¹;
-
Solução de carbonato de sódio (Na₂CO₃, CAS 497-19-8) a 20% (m/v);
-
Soluções padrão de ácido gálico (C₆H₂(OH)₃COOH, CAS 149-91-7):
Dissolver 0,250 g de ácido gálico em 5 mL de etanol e diluir para 50 mL com água em balão volumétrico. Diluir, em balão volumétrico, alíquotas de 200 μL, 400 μL, 600 μL, 800 μL e 1000 μL desta solução para 10 mL, obtendo-se soluções padrão de 100 mg·L⁻¹, 200 mg·L⁻¹, 300 mg·L⁻¹, 400 mg·L⁻¹ e 500 mg·L⁻¹, respectivamente. A solução estoque é estável por até 2 semanas se estocada sob refrigeração a 4°C–8°C.
¶ Preparo da amostra
Agitar a embalagem para homogeneização da amostra.
¶ Procedimento de análise
-
Em balão volumétrico de 25 mL, pesar aproximadamente 250 mg da amostra, completando para o volume com metanol.
-
Adicionar uma alíquota de 100 μL da amostra diluída e de cada uma das soluções padrão a diferentes balões volumétricos de 10 mL;
-
Adicionar 7 mL de água e 0,5 mL do reagente de Folin-Ciocalteau;
-
Agitar e incubar a temperatura ambiente entre 1 e não mais que 8 min;
-
Adicionar 1,5 mL de solução de carbonato de sódio a 20% (m/v);
-
Completar o volume com água e deixar em repouso por 60 min a temperatura ambiente;
-
Transferir para cubeta do espectrofotômetro e efetuar leitura da amostra e das soluções padrão a 760 nm, utilizando um branco dos reagentes como referência, registrando os valores obtidos.
Analisar as amostras em duplicata, obtendo uma nova curva de calibração a cada batelada.
¶ Cálculo e expressão dos resultados
A partir dos valores de absorbância e concentração dos padrões em mg·L⁻¹, obter pelo método dos mínimos quadrados a equação da curva de calibração. A partir desta equação, obter a concentração de compostos fenólicos na amostra diluída, utilizando a equação abaixo para determinar a concentração na amostra:
onde:
W = Concentração de compostos fenólicos expressos em ácido gálico, obtida da curva de calibração;
Vb = volume do balão volumétrico utilizado na diluição inicial, em mL (25 mL);
m = massa da amostra, em mg;
0,1 = fator de conversão de g/kg para g/100 g.
Expressar como resultado a média da duplicata com uma casa decimal.
¶ Bibliografia
Ricardo G. Woisky e Antonio Salatino. Analysis of propolis: some parameters and procedures for chemical quality control. Journal of Apicultural Research. Vol. 37(2), p. 99-105, 1998.
Andrew L. Waterhouse. Determination of Total Phenolics, in: Current Protocols in Food Analytical Chemistry, I1.1.1-I1.1.8, Wiley, 2001.
¶ Compostos flavonoides em própolis
Determinar o teor de flavonoides totais utilizando o método descrito na norma ABNT NBR 16956-4, reportando o resultado obtido em “g/100 g” com duas casas decimais.
¶ Compostos flavonoides em extrato de própolis
¶ Princípio
Os flavonoides solubilizados em metanol podem combinar-se com o reagente cromogênico para formar uma substância colorida, com absorção máxima próxima ao comprimento de onda de 425 nm.
¶ Campo de aplicação
Este método é aplicável a extrato de própolis.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,0001 g;
-
Balões volumétricos de 10 mL, 25 mL, 50 mL e 100 mL;
-
Espectrofotômetro de absorção molecular capaz de efetuar leituras em 425 nm;
-
Micropipetador de 100 μL–1000 μL.
¶ Reagentes e soluções
-
Metanol (CH₃OH, CAS 67-56-1);
-
Solução de cloreto de alumínio (AlCl₃, CAS 7446-70-0) a 5% em metanol;
-
Solução padrão de quercetina (C₁₅H₁₀O₇, CAS 117-39-5) a 300 mg·L⁻¹ em metanol.
¶ Preparo da amostra
Agitar a embalagem para homogeneização da amostra.
¶ Procedimento de análise
-
Em balão volumétrico de 25 mL, pesar aproximadamente 250 mg da amostra, completando para o volume com metanol.
-
Em balões volumétricos de 25 mL, adicionar alíquotas de 200 μL, 300 μL, 400 μL, 500 μL e 600 μL da solução padrão de quercetina e 2 mL da amostra diluída.
-
Adicionar, a cada balão, aproximadamente 15 mL de metanol e 500 μL da solução de cloreto de alumínio a 5% em metanol,
-
Completar o volume com metanol, agitar e aguardar 30 min com o balão protegido da luz;
-
Determinar a absorbância dos padrões e da amostra em 425 nm em cubeta de caminho óptico de 1 cm utilizando como referência um branco dos reagentes.
Analisar as amostras em duplicata, obtendo uma nova curva de calibração a cada batelada.
¶ Cálculo e expressão dos resultados
A partir dos valores de absorbância e concentração dos padrões em mg l⁻¹, obter pelo método dos mínimos quadrados a equação da curva de calibração. A partir desta equação, obter a concentração de compostos flavonoides na amostra diluída, utilizando a equação abaixo para determinar a concentração na amostra:
onde:
W = Concentração de compostos fenólicos expressos em quercetina, obtida da curva de calibração;
Vb = volume do balão volumétrico utilizado na reação, em mL (25 mL);
Vdi = volume do balão volumétrico utilizado na diluição inicial, em mL (25 mL);
Va = volume da alíquota adicionada ao balão da medida, em mL (2 mL);
m = massa da amostra, em mg;
0,1 = fator de conversão de g/kg para g/100 g.
¶ Bibliografia
Associação Brasileira de Normas Técnicas. ABNT NBR 16956-4:2021 - Apicultura - Própolis - Parte 4: Determinação de flavonoides totais. São Paulo: 2021.
¶ Hidroximetilfurfural
Determinar o teor de hidroximetilfurfural (HMF) utilizando o método descrito nas normas ABNT NBR 15714-9 ou AOAC 980.23, reportando o resultado obtido em “mg de HMF/kg” com uma casa decimal.
¶ Índice de acidez, ésteres e de relação
¶ Princípio
Fundamenta-se na neutralização até o ponto de equivalência dos ácidos existentes em 1 g de cera para obtenção do Índice de Acidez. A saponificação total dos glicerídeos e ésteres e posterior titulação dos ácidos graxos resultantes fornece o Índice de Ésteres.
¶ Campo de aplicação
Cera de abelha.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Bureta de 10 mL com resolução mínima de 0,1 mL;
-
Condensador de refluxo;
-
Erlenmeyer de 250 mL com junta esmerilhada;
-
Pipetas volumétricas de 25 mL e 50 mL;
-
Placa aquecedora.
¶ Reagentes e soluções
-
Etanol (C₂H₅OH, CAS 64-17-5) neutralizado com hidróxido de potássio (KOH, CAS 1310-58-3) a 0,5 mol·L⁻¹ e fenolftaleína (C₂₀H₁₄O₄, CAS 77-09-8) como indicador;
-
Solução de ácido clorídrico (HCl, CAS 7647-01-0) a 0,5 mol·L⁻¹;
-
Solução alcoólica de fenolftaleína a 1% (m/v);
-
Solução etanólica de hidróxido de potássio (KOH) a 0,5 mol·L⁻¹.
¶ Preparo da amostra
Picar a amostra em pequenas porções e armazenar em recipiente fechado.
¶ Procedimento de análise
-
Pesar cerca de 3 g de amostra em Erlenmeyer;
-
Adicionar 25 mL de etanol neutralizado e aquecer até dissolução da amostra;
-
Adicionar 5 gotas de solução alcoólica de fenolftaleína a 1% (m/v) e titular com solução etanólica de hidróxido de potássio 0,5 mol·L⁻¹ até róseo persistente durante 30 s;
-
Utilizar o volume gasto de solução hidróxido de potássio 0,5 mol·L⁻¹ (VKOH) para determinação do Índice de Acidez;
-
Adicionar ao erlenmeyer exatamente 25 mL de solução etanólica de hidróxido de potássio 0,5 mol·L⁻¹ e 50 mL de álcool etílico neutralizado;
-
Deixar em refluxo por 4 h;
-
Titular, sob aquecimento, com solução de ácido clorídrico 0,5 mol·L⁻¹ até descoramento;
-
Utilizar o volume gasto de ácido clorídrico 0,5 mol·L⁻¹ (VHCl) para determinação do índice de ésteres;
-
Efetuar prova em branco, anotando o volume de ácido clorídrico 0,5 mol·L⁻¹ gasto no branco (Vbranco).
¶ Cálculo e expressão dos resultados
onde:
VKOH = volume de solução etanólica de hidróxido de potássio 0,5 mol·L⁻¹ gasto na titulação, em mL;
VHCl = volume de solução de ácido clorídrico 0,5 mol·L⁻¹ gasto na titulação, em mL;
Vbranco = volume de solução de ácido clorídrico 0,5 mol·L⁻¹ gasto na titulação do branco, em mL;
m = massa da amostra, em g;
f = fator de correção da solução etanólica de hidróxido de potássio 0,5 mol·L⁻¹;
f′ = fator de correção da solução de ácido clorídrico 0,5 mol·L⁻¹.
Expressar os resultados obtidos com uma casa decimal.
¶ Bibliografia
FAO JECFA. Combined Compendium for Food Additive Specifications - Analytical methods, test procedures and laboratory solutions used by and referenced in the food additive specifications, Monographs No. 1, Vol. 4. Roma: 2006.
United States Pharmacopeial Convention. Food Chemicals Codex. 4th ed. Washington: National Academy Press, 1996.
¶ Insolúveis
Utilizar o método descrito na norma ABNT NBR 15714-5, expressando o resultado final em “g/100 g” com uma casa decimal.
¶ Massa mecânica (insolúveis no etanol), cera e solúveis no etanol
Utilizar o método descrito na norma ABNT NBR 16956-1 para a obtenção do teor de cera, teor de sólidos solúveis e a massa mecânica (teor de resíduos insolúveis), expressando o resultado final em “g/100 g” com uma casa decimal.
¶ Perda por dessecação em própolis
Determinar a perda por dessecação utilizando o método descrito na norma ABNT NBR 16956-2, reportando o resultado obtido em “g/100 g” com uma casa decimal.
¶ Extrato seco
¶ Princípio
Secagem de uma alíquota da amostra, a uma temperatura determinada, até massa constante e pesagem para determinação do extrato seco.
¶ Campo de aplicação
Este método aplica-se a extrato de própolis.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,0001 g;
-
Banho-maria;
-
Cápsulas de alumínio, vidro, aço inoxidável ou níquel, com cerca de 25 mm de profundidade e diâmetro de aproximadamente 50 mm;
-
Estufa com circulação de ar a (102 ± 2)°C.
¶ Reagentes e soluções
Não aplicável.
¶ Preparo da amostra
Homogeneizar a amostra em seu recipiente.
¶ Procedimento de análise
-
Aquecer a cápsula em uma estufa a (102 ± 2)°C por 1 h, transferir para dessecador, esfriar até a temperatura ambiente e pesar (m₀);
-
Transferir 5 mL da amostra para cápsula;
-
Levar o conjunto ao banho-maria e secar sob vapor por 30 min;
-
Transferir a cápsula para estufa a (102 ± 2)°C, por 1 h;
-
Transferir para dessecador e esfriar por aproximadamente 30 min;
-
Pesar e anotar a massa;
-
Repetir o procedimento de 4 a 6, utilizando tempo de secagem de 30 min, até que a variação entre duas pesagens consecutivas seja inferior a 5 mg ou que a amostra ganhe massa, devendo-se considerar a menor massa obtida como a massa final (m₁).
¶ Cálculo e expressão dos resultados
onde:
m₀ = massa da cápsula, em gramas;
5 = volume da amostra, em mililitros;
m₁ = massa da cápsula + amostra seca, em gramas.
Expressar como resultado a média das duplicatas com duas casas decimais.
¶ Bibliografia
Ricardo G. Woisky e Antonio Salatino. Analysis of propolis: some parameters and procedures for chemical quality control. Journal of Apicultural Research. Vol. 37(2), p. 99-105, 1998.
International Organization for Standardization. 6731:2010 – Milk, cream and evaporated milk – Determination of total solids content (Reference method). Genebra: 2010.
Associação Brasileira de Normas Técnicas. ABNT NBR 16956-2:2021 - Apicultura - Própolis - Parte 2: Determinação da perda por dessecação e teor de umidade. São Paulo: 2021.
¶ Ponto de fusão
¶ Princípio
Baseia-se na determinação do ponto de fusão de uma amostra submetida a aquecimento controlado por meio de equipamento específico.
¶ Campo de aplicação
Este método é aplicável a cera de abelhas.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 1 g;
-
Béquer de 50 mL;
-
Funil de vidro;
-
Material descrito na norma ISO 6321;
-
Papel de filtro.
¶ Reagentes e soluções
Não aplicável.
¶ Procedimento de análise
-
Pesar cerca de 15 g da amostra em um béquer e fundir em estufa a 70°C;
-
Filtrar com papel de filtro qualitativo;
-
Obter o ponto de fusão de acordo com o método A descrito na norma ISO 6321.
¶ Expressão dos resultados
Reportar o valor obtido com uma casa decimal.
¶ Bibliografia
BRASIL. Ministério da Agricultura. Secretaria Nacional de Defesa Agropecuária. Laboratório Nacional de Referência Animal. Métodos analíticos oficiais para controle de produtos de origem animal e seus ingredientes: II – Métodos físicos e químicos. Brasília: 1981.
International Organization for Standardization. 6321:2002 – Animal and vegetable fats and oils – Determination of melting point in open capillary tubes (slip point). Genebra: 2002.
¶ Ponto de saponificação turva
¶ Princípio
Baseia-se na medição da temperatura em que os ácidos graxos iniciam a solidificação, após saponificação.
¶ Campo de aplicação
Este método aplica-se a cera de abelha.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Balão de fundo chato de 100 mL;
-
Banho-maria a 80°C;
-
Bureta de 50 mL;
-
Condensador de refluxo;
-
Termômetro com resolução mínima de 1°C.
¶ Reagentes e soluções
-
Solução de saponificação:
Dissolver 4.0 g de hidróxido de potássio (KOH, CAS 1310-58-3) em 90 mL de etanol (C₂H₅OH, CAS 64-17-5) livre de aldeídos mantido a aproximadamente 15°C durante a dissolução. Deixar atingir a temperatura ambiente, transferir para balão volumétrico de 100 mL e completar o volume.
¶ Procedimento de análise
-
Pesar cerca de 15 g da amostra em um béquer e fundir em estufa a 70°C;
-
Filtrar com papel de filtro qualitativo;
-
Pesar (3,0 ± 0,1) g da amostra filtrada e passar para balão de fundo chato de 100 mL;
-
Adicionar 30 mL de solução de saponificação;
-
Acoplar o condensador e refluxar a mistura por 2 h;
-
Transcorrido o tempo, desconectar o condensador;
-
Colocar, dentro do balão, um termômetro;
-
Girar suavemente o balão enquanto a temperatura da solução decresce. A temperatura em que se notar uma turvação da solução ou formação de pequenos glóbulos será considerado o ponto de saponificação turva.
Para maior precisão da leitura, colocar embaixo do balão e colado neste, um cartão tendo impresso letras negras com altura de 1 cm. Quando as letras observadas através do balão tornam-se de difícil visibilidade, será esta a temperatura considerada como ponto de turvação.
¶ Expressão dos resultados
Reportar a temperatura do ponto de saponificação turva como um número inteiro.
¶ Bibliografia
United States Pharmacopeial Convention. Food Chemicals Codex. 4th ed. Washington: National Academy Press, 1996.
¶ Resíduo mineral fixo/Cinzas
¶ Mel
Utilizar o método descrito na norma ABNT NBR 15714-3, expressando o resultado obtido em “g/100 g” com uma casa decimal.
¶ Própolis
Utilizar o método descrito na norma ABNT NBR 16956-3, expressando o resultado obtido em “g/100 g” com uma casa decimal.
¶ Sacarose
Determinar o teor de “Sacarose” em “g/100 g” pelo método AOAC 977.20 ou pelo método descrito na Seção Lactose, glicose, frutose e sacarose por cromatografia iônica, reportando o valor obtido com uma casa decimal.
¶ Teor alcoólico em extrato de própolis
Com o auxílio do método descrito na norma AOAC 983.12, determinar o teor alcoólico na amostra, expressando o resultado em “°GL (v/v)” com uma casa decimal.
Poderão ser utilizados equipamentos automatizados de destilação para a execução do método.
¶ Teste para adulteração por açúcares C-4
Determinar a quantidade aparente de açúcares C-4 por meio do Método II da norma AOAC 998.12. Reportar positivo se o valor obtido for superior a 7%.
¶ Teste para cera de carnaúba
¶ Princípio
Baseia-se nas diferenças entre os pontos de cristalização em álcool n-butílico da cera de abelha e da cera de carnaúba.
¶ Campo de aplicação
Este ensaio aplica-se a cera de abelhas.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Banho-maria;
-
Microscópio ou lente de aumento;
-
Tubos de ensaio.
¶ Reagentes e soluções
- 1-butanol (C₄H₉OH, CAS 71-36-3).
¶ Procedimento
-
Pesar aproximadamente 50 mg de amostra em tubo de ensaio;
-
Adicionar ao tubo 10 mL de álcool n-butílico;
-
Aquecer, sob agitação, o tubo de ensaio em banho de água em ebulição até completa dissolução;
-
Transferir o tubo de ensaio para banho a 60°C e deixá-lo em repouso;
-
Observar os cristais formados.
¶ Expressão dos resultados
Reportar negativo se os cristais formados parecerem agulhas soltas ou aglomeradas, sem presença de massas amorfas sob o microscópio ou lente de aumento.
¶ Bibliografia
United States Pharmacopeial Convention. Food Chemicals Codex. 4th ed. Washington: National Academy Press, 1996.
¶ Teste para cera japonesa, resinas e gorduras
¶ Princípio
Baseia-se nas diferenças de solubilidade dos produtos de saponificação da cera de abelha e de suas impurezas.
¶ Campo de aplicação
Este ensaio aplica-se a cera de abelhas.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Erlenmeyer de 125 mL;
-
Papel de filtro qualitativo.
¶ Reagentes e soluções
-
Ácido clorídrico (HCl, CAS 7647-01-0) 3.5 mol·L⁻¹;
-
Papel de pH ou solução a 1% de fenolftaleína (C₂₀H₁₄O₄, CAS 77-09-8);
-
Solução de hidróxido de sódio (NaOH, CAS 1310-73-2) a 3.5 mol·L⁻¹.
¶ Procedimento
-
Pesar (1,0 ± 0,1) g de amostra em erlenmeyer;
-
Adicionar 35 mL da solução de hidróxido de sódio;
-
Manter sob ebulição por 30 min, mantendo o volume constante por adição ocasional de água;
-
Após este período, ocorre a separação entre a cera e a parte líquida, a qual deve manter-se límpida. Esfriar e filtrar em papel qualitativo;
-
Acidificar o filtrado com solução de ácido clorídrico;
-
Observar se há formação de precipitados.
¶ Expressão dos resultados
Reportar positivo se houver formação de precipitados após acidificação.
¶ Bibliografia
United States Pharmacopeial Convention. Food Chemicals Codex. 4th ed. Washington: National Academy Press, 1996.
¶ Umidade em mel
Utilizar o método B da norma AOAC 969.38 ou a norma ABNT NBR 15714-2, reportando o valor obtido em “g/100 g” com uma casa decimal.
¶ Ovos e derivados
¶ Lipídios
Determinar o teor lipídico da amostra por meio da norma AOAC 925.32, reportando o resultado com duas casas decimais em “g/100 g”.
¶ pH
Utilizar o método descrito na Seção Leite e produtos lácteos - pH.
¶ Proteína
Multiplicar o teor de nitrogênio na amostra, determinado em acordo com a norma ISO 1871, pelo fator de conversão 6,25, expressando o resultado obtido “em g/100 g” com duas casas decimais. Alternativamente, utilizar a norma AOAC 992.15.
¶ Resíduo mineral fixo
¶ Princípio
Fundamenta-se na eliminação da matéria orgânica e inorgânica volátil à temperatura de 550°C. O produto obtido é denominado de resíduo mineral fixo ou cinzas.
¶ Campo de aplicação
Ovo líquido e ovo desidratado.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,0001 g;
-
Cadinho de porcelana, quartzo, metal ou qualquer outro metal inerte nas condições do ensaio com diâmetro mínimo de 60 mm e paredes inclinadas com altura mínima de 25 mm;
-
Dessecador com sílica gel ou cloreto de cálcio anidro;
-
Forno tipo mufla capaz de atingir e manter a temperatura de 550 ± 25°C;
-
Pinça metálica.
¶ Reagentes e soluções
- Peróxido de hidrogênio (H₂O₂, CAS 7722-84-1) a 30% (v/v).
¶ Preparo da amostra
Homogenizar a amostra agitando e invertendo o recipiente ou embalagem 5 ou 6 vezes.
¶ Procedimento de análise
-
Aquecer o cadinho em forno mufla a 550°C durante 20 min, esfriar em dessecador até temperatura ambiente e pesar (m₀);
-
Pesar entre 2 g e 3 g da amostra homogeneizada no cadinho (m₁);
-
Colocar o cadinho no forno mufla e aumentar gradualmente a temperatura até (550 ± 25)°C, mantendo-o nesta temperatura por tempo suficiente para obtenção de cinzas claras (aproximadamente 4 h);
-
Retirar o cadinho para dessecador e esfriar até temperatura ambiente;
-
Observar a coloração das cinzas. Se obtidas cinzas de coloração negra, adicionar algumas gotas de peróxido de hidrogênio a 30% e repetir as etapas de incineração. Se as cinzas possuem coloração entre branco e cinza claro, pesar o conjunto cinzas+cadinho (m₂);
¶ Cálculo e expressão dos resultados
onde:
m₀ = massa do cadinho, em g;
m₁ = massa da amostra, em g;
m₂ = massa do cadinho + cinzas, em g.
Reportar a média aritmética de duas determinações com uma casa decimal.
¶ Bibliografia
International Organization for Standardization. ISO 936:1998 – Meat and meat products - Determination of total ash. Genebra: 1998.
¶ Sólidos totais
Determinar o teor de sólidos totais na amostra por meio da norma AOAC 925.30, reportando o resultado com duas casas decimais em “g/100 g”.
¶ Pescados e subprodutos da pesca
¶ Preparo de amostras
Instruções específicas, requeridas e descritas nos métodos, também devem ser observadas. Caso a análise não se inicie imediatamente, manter a amostra preparada sob refrigeração em recipiente hermeticamente fechado. Para prevenir perda de água durante o preparo, utilize porções tão grandes quanto possível. Preparar o pescado como para consumo, descartando ossos e outras partes não comestíveis, salvo instrução contrária presente na metodologia analítica.
¶ Peixes in natura
Limpar o peixe removendo, se presentes, escamas e vísceras. Filetar o peixe de ambos os lados. Para peixes pequenos (inferiores a 15 cm), utilizar entre 5 e 10 unidades.
Para peixes grandes, a cada 3 peixes cortar seção transversal de aproximadamente 2,5 cm logo após a nadadeira peitoral, uma seção entre o primeiro corte e o ânus e uma seção logo após o ânus. Descartar pele e ossos.
Em moinho: Moer rapidamente por três vezes em moedor com abertura de 3 mm, misturando após cada moagem.
Em processador de alimentos: Cortar a amostra em pedaços de aproximadamente 5 cm e transferir, juntamente com qualquer líquido, para o recipiente do equipamento. Processar por 30 s, raspar as laterais do recipiente com espátula e reincorporar à porção principal da amostra, assim como qualquer material preso nas lâminas. Processar por 30 s adicionais. Repetir este processo por duas vezes. Se utilizado equipamento que realize a raspagem simultaneamente ao processamento, pode-se efetuar o preparo em uma única etapa de aproximadamente um minuto.
¶ Conservas e produtos enlatados
Preparar todo o conteúdo da lata em moinho ou processador de alimentos como em Preparo de amostras- Peixes in natura.
¶ Produtos salgados, salgados secos e defumados
Remover, se presentes, ossos e o sal aderido à superfície. Preparar peixes inteiros como em Preparo de amostras- Peixes in natura. Para produtos em pedaços, homogeneizar em moinho ou processador de alimentos como em Preparo de amostras- Peixes in natura.
Bacalhau espalmado e similares (p. ex. Saithe) devem ser amostrados e analisados de acordo com o método cross section descrito no Codex Stan 167 (item 5.25).
¶ Moluscos bivalves e cefalópodes
Cortar porções da musculatura e homogeneizar em moinho ou processador de alimentos como em Preparo de amostras- Peixes in natura.
¶ Crustáceos
Remover a carapaça, cortar porções da musculatura e homogeneizar em moinho ou processador de alimentos como em Preparo de amostras- Peixes in natura. Para camarões e lagostas, não utilizar a cabeça.
¶ Bibliografia
AOAC International. Official Methods of Analysis of AOAC International, Official Method 937.07. 21 ed. Rockville: 2019.
¶ Amido
Utilizar o método descrito na Seção Carnes e produtos cárneos - Amido e carboidratos totais.
¶ Anidrido sulfuroso e sulfitos
Utilizando o método descrito na norma AOAC 990.28, determinar o teor de sulfitos na porção edível do pescado, expressando o resultado em “mg de SO₂/kg” como um número inteiro.
¶ Bases voláteis totais
¶ Princípio
As bases voláteis são extraídas da amostra com uma solução de ácido perclórico. Após alcalinização, o extrato é submetido a destilação por arraste de vapor, sendo os componentes de básicos voláteis absorvidos pelo ácido receptor. A concentração de BVT é determinada por titulação das bases absorvidas.
¶ Campo de aplicação
Este método aplica-se a pescados e produtos a base de pescado.
¶ Materiais e equipamentos
-
Balança analítica com resolução mínima de 0,1 g;
-
Bureta com resolução mínima de 0,05 mL, ou bureta digital que atenda ao critério de resolução;
-
Equipamento para destilação por arraste de vapor;
-
Erlenmeyers;
-
Funil de vidro;
-
Homogeneizador tipo Turrax com velocidade igual ou superior a de 15000 RPM;
-
Papel de filtro;
-
Proveta de 25 mL;
¶ Reagentes e soluções
-
Antiespumante a base de silicone;
-
Solução de ácido bórico (H₃BO₃, CAS 10043-35-3) a 30 g·L⁻¹;
-
Solução de ácido perclórico (ClHO₄, CAS 7601-90-3) a 60 g·L⁻¹;
-
Solução de hidróxido de sódio (NaOH, CAS 1310-73-2) a 200 g·L⁻¹;
-
Solução de fenolftaleína (C₂₀H₁₄O₄, CAS 77-09-8) a 10 g·L⁻¹;
-
Solução indicadora de Tashiro:
Dissolver 0,2 g de vermelho de metila (C₁₅H₁₅N₃O₂, CAS 493-52-7) e 0,1 g de azul de metileno (C₁₆H₁₈N₃S·Cl , CAS 61-73-4) em 100 mL de etanol (C₂H₅OH, CAS 64-17-5) a 95%.
-
Solução padronizada de ácido clorídrico (HCl, CAS 7647-01-0) a 0,1 mol·L⁻¹;
¶ Preparo da amostra
-
Pescado congelado:
Remover, se presente, a água de recobrimento (“glaciamento”) da amostra utilizando água corrente. Retirar porções musculares da amostra ainda congelada, triturar e homogeneizar. Analisar imediatamente.
-
Pescado resfriado:
Retirar porções musculares da amostra, triturar e homogeneizar. Analisar imediatamente.
¶ Procedimento de análise
-
Pesar (10,0 ± 0,1) g de amostra triturada para erlenmeyer;
-
Adicionar 90 mL de a 60 g·L⁻¹;
-
Homogeneizar em Turrax por 2 min;
-
Filtrar. O filtrado, se mantido refrigerado, pode ser analisado em até 7 dias;
-
Colocar 50 mL do filtrado no dispositivo de destilação por arraste de vapor;
-
Adicionar ao filtrado cerca de 5 gotas de solução de fenolftaleína. Se necessário, adicionar algumas gotas de antiespumante;
-
Adicionar 6,5 mL de NaOH a 200 g·L⁻¹. A alcalinização é verificada mediante a mudança de cor para rosa;
-
Regular o equipamento de modo a obter cerca de 100 mL de destilado em 10 min. O destilado é recolhido em recipiente com 100 mL de solução de ácido bórico;
-
Após 10 min, recolher o destilado e adicionar ao mesmo algumas gotas do indicador de Tashiro;
-
Titular com solução de ácido clorídrico até a viragem do indicador. Opcionalmente, pode-se utilizar como ponto final da titulação o pH de (5,0 ± 0,1).
O ensaio deve ser conduzido em duplicata. Os resultados obtidos são considerados corretos se a diferença entre as duplicatas não exceder 2 mg de N/100 g. Deve ser realizada uma prova em branco, substituindo-se os 50 mL do filtrado por 50 mL de solução de ácido perclórico.
O equipamento para destilação por arraste de vapor deve ser verificado mediante a destilação de soluções de sulfato de amônio ((NH₄)₂SO₄, CAS 7783-20-2), tolerando-se uma recuperação entre 99% e 101%.
Equipamentos automatizados podem ser utilizados, desde que baseados nos mesmos princípios e satisfaçam a condição de verificação.
¶ Expressão dos resultados
onde:
V = volume de solução de 0,1 mol·L⁻¹ gasto na titulação da amostra, em mL;
Vb = volume de solução de 0,1 mol·L⁻¹ gasto na titulação do branco, em mL;
f = fator de correção da solução de 0,1 mol·L⁻¹;
0,1 = molaridade de solução de , em mol·L⁻¹;
14 = massa molar do nitrogênio, em g mol⁻¹;
2 = fator de diluição;
m = massa da amostra, em g.
Reportar a média aritmética das duplicatas como um número inteiro.
¶ Bibliografia
Jornal Oficial das Comunidades Europeias, nº L97 de 29/04/1995, p. 84-87.
¶ Cloreto de sódio (NaCl)
¶ Princípio
O sal é extraído com água da amostra. Após precipitação das proteínas, a concentração de cloretos é determinada por titulação de uma alíquota da solução com nitrato de prata padronizado (método de Mohr) e calculado como cloreto de sódio.
¶ Campo de aplicação
Este método aplica-se a pescado, produtos a base de pescado, carne e produtos cárneos.
¶ Materiais e equipamentos
-
Agitador magnético;
-
Balança analítica com resolução mínima de 0,01 g;
-
Balão volumétrico de 250 mL;
-
Bureta;
-
Erlenmeyers;
-
Funil de vidro;
-
Papel de filtro.
¶ Reagentes e soluções
-
Solução de Carrez I:
Dissolver 10,6 g de hexacianoferrato(II) de potássio (K₄[Fe(CN)₆]·3H₂O, CAS 14459-95-1) em água, transferir para um balão volumétrico de 100 mL e completar para o volume.
-
Solução de Carrez II:
A um balão volumétrico de 100 mL, adicionar 21,9 g de acetato de zinco dihidratado (CH₃COO)₂Zn·2H₂O, CAS 5970-45-6) e 3,2 mL de ácido acético (CH3COOH, CAS 64-19-7). Diluir para o volume com água.
-
Solução de cromato de potássio (K₂CrO₄, CAS 7789-00-6) a 5% m/v;
-
Solução de fenolftaleína (C₂₀H₁₄O₄, CAS 77-09-8) a 10 g·L⁻¹;
-
Solução de hidróxido de sódio (NaOH, CAS 1310-73-2) a 0,1 mol·L⁻¹;
-
Solução padrão de nitrato de prata (AgNO₃, CAS 7761-88-8) a 0,1 mol·L⁻¹.
¶ Preparo da amostra
Retirar porções musculares da amostra, triturar e homogeneizar.
¶ Procedimento de análise
-
Pesar 5 g de amostra para balão volumétrico de 250 mL;
-
Adicionar aproximadamente 100 mL de água e agitar vigorosamente;
-
Adicionar 5 mL de solução de Carrez I e agitar;
-
Adicionar 5 mL de solução de Carrez II e agitar;
-
Completar o volume do balão com água;
-
Após nova agitação, filtrar o conteúdo do balão.
-
Transferir um alíquota do filtrado para Erlenmeyer, adicionando duas gotas de solução de fenolftaleína;
-
Adicionar solução de hidróxido de sódio lentamente até o surgimento de uma coloração rósea;
-
Adicionar cerca de 100 mL de água e 1 mL de solução de cromato de potássio;
-
Titular com solução de nitrato de prata, sob agitação constante, até mudança persistente da coloração da solução para vermelho-tijolo claro.
O ensaio deve ser conduzido em duplicata. Deve ser realizada uma prova em branco dos reagentes.
¶ Expressão dos resultados
onde:
V = volume de solução de 0,1 mol·L⁻¹ gasto na titulação da amostra, em mL;
Vb = volume de solução de 0,1 mol·L⁻¹ gasto na titulação do branco, em mL;
f = fator de correção da solução de 0,1 mol·L⁻¹;
0,1 = molaridade de solução de , em mol·L⁻¹;
58,44 = massa molar do , em g·mol⁻¹;
250 = volume do balão volumétrico;
A = alíquota tomada, em mL;
m = massa da amostra, em g.
Reportar o teor de sal (NaCl) como a média aritmética das duplicatas em “g de NaCl/100 g” com uma casa decimal.
¶ Bibliografia
Codex Alimentarius. Codex Standard for Salted Fish and Dried Salted Fish of the Gadidae Family of Fishes – Codex Stan 167-1989. Roma: FAO/WHO, 2016.
¶ Corantes artificiais
Utilizando a metodologia descrita na norma NMKL 130, reportar “presença de nome comum (código INS)” para cada corante identificado. Reportar “presença corante hidrossolúvel não identificado” caso seja detectado um corante sem correlação com os corantes testados no método. Deve-se testar, em adição aos corantes descritos na norma, a presença do corante carmoisina (INS 122).
¶ Desglaciamento
¶ Princípio
O método baseia-se na remoção em condições controladas do glaciamento da amostra para determinação do percentual de glaciamento.
¶ Campo de aplicação
Pescados congelados glaciados.
¶ Materiais e equipamentos
-
Balança com resolução mínima de 0,1 g;
-
Cronômetro;
-
Peneira com malha de 2,4 mm em aço inoxidável;
-
Recipiente paralelepipédico com um volume superior a 10 vezes o peso bruto da amostra;
-
Termômetro com precisão de 0,1°C, abrangendo a faixa de 15°C a 25°C;
-
Toalha de tecido ou papel.
¶ Reagentes e soluções
Não aplicável.
¶ Preparo da amostra
A temperatura de armazenagem da amostra não pode exceder −8°C. No momento do ensaio, a amostra deve estar a uma temperatura inferior a −12°C.
¶ Procedimento de análise
-
Pesar a amostra com embalagem e isenta de gelo exterior, obtendo-se o peso bruto (PB) da amostra;
-
Pesar a embalagem e/ou invólucro totalmente limpos e sem resíduos, obtendo-se assim o valor do peso da embalagem (PE);
-
Com o produto já sem embalagem, acomodá-lo na peneira e submergir o conjunto em um recipiente contendo um volume aproximado de água de 10 vezes o peso da amostra, observando o volume mínimo de 10 L. O banho deve estar a uma temperatura de (20 ± 2)°C;
-
Manter o conjunto peneira mais produto submerso até a percepção tátil de que todo o glaciamento foi removido, evitando-se o descongelamento substancial da amostra;
-
Retirar o conjunto peneira mais produto e deixar escorrer por (60 ± 10) s. Para facilitar a drenagem, a peneira deverá permanecer inclinada, preferencialmente em um ângulo entre 15° e 17°. A água aderida à superfície da amostra pode ser removida com o auxílio de toalhas de papel ou tecido, evitando-se pressionar a amostra;
-
Pesar a amostra desglaciada, determinando o peso do produto desglaciado (Pd);
-
Repetir este procedimento para as 5 amostras restantes.
Para amostras de camarão é recomendável que a peneira seja pesada antes do banho e a amostra desglaciada pesada em conjunto com a mesma, subtraindo-se o peso da peneira do peso obtido para obtenção do Pd da amostra.
A utilização da peneira para imersão no banho é opcional para amostras compostas por uma peça única.
¶ Cálculo e expressão dos resultados
- Determinar o peso do produto glaciado (Pg) para cada amostra subtraindo-se do peso bruto o peso da embalagem correspondente:
-
Determinar a média aritmética (Pd) e desvio padrão (sPd) dos Pd.
-
Determinar o percentual de glaciamento (%G) utilizando a fórmula:
- Calcular o peso do produto desglaciado a ser considerado para inspeção (Pdc), “peso desglaciado + tolerância”, através da equação Pdc = Pd + 1,646·sPd.
Para pescados pré-medidos de peso declarado uniforme, reportar:
-
o “peso glaciado” (Pg), como um número inteiro;
-
o “peso desglaciado + tolerância” (Pdc), como um número inteiro;
-
o “percentual de glaciamento” (%G), em % como um número inteiro.
Adicionalmente, informar no campo “Observações” do certificado/relatório o peso líquido declarado pelo fabricante.
Para pescados pré-embalados com conteúdo nominal desigual, reportar:
- o “percentual de glaciamento” (%G), em % como um número inteiro.
¶ Bibliografia
Codex Alimentarius. Codex Standard for Quick Frozen Lobsters – Codex Stan 95 rev. 2. Roma: FAO/WHO, 2004.
Codex Alimentarius. Codex Standard for Quick Frozen Shrimps or Prawns – Codex Stan 92 rev. 1. Roma: FAO/WHO, 1995.
Codex Alimentarius. Codex Standard for Quick Frozen Blocks os Fish Fillets, Minced Fish Flesh and Mixtures of Filets and Minced Fish Flesh – Codex Stan 165 rev. 1. Roma: FAO/WHO, 1995.
AOAC International. Official Methods of Analysis of AOAC International, Official Method 963.18. 20 ed. Rockville: 2016.
Codex Alimentarius. General Guidelines on Sampling – CAC/GL 50. Roma: FAO/WHO, 2004.
¶ Detecção de formaldeído
Utilizar o método B da norma AOAC 931.08. Reportar “positivo” caso seja detectada a presença de formaldeído na amostra e “negativo” em caso contrário.
¶ Detecção de polifosfatos
¶ Princípio
Os ânions polifosfatos são extraídos em água, separados em coluna iônica e derivatizados com auxílio de reagente de molibdato de amônio para detecção no visível.
¶ Campo de aplicação
Pescados congelado, pescado congelado com água de recobrimento contendo polifosfatos, pescado com água de recobrimento sem adição de polifosfatos, carne e produtos cárneos congelados.
¶ Materiais e equipamentos
-
Aparelho para filtração de solventes para cromatografia líquida dotado de membrana de 45 μm;
-
Balança analítica com resolução mínima de 0,001 g;
-
Balança semi-analítica com resolução mínima de 0,1 g;
-
Balão volumétrico de 1 L;
-
Centrífuga capaz de gerar uma RCF de 3000g ou superior;
-
Homogeneizador tipo Turrax com velocidade igual ou superior a de 15000 RPM;
-
Proveta de 25 mL;
-
Seringas acopladas a filtro de membrana de 45 μm;
-
Sistema de cromatografia líquida de alta eficiência (CLAE, HPLC), dotado de bomba isocrática, forno de reação pós-coluna capaz de manter uma temperatura de (115 ± 2)°C ou superior, bomba auxiliar para derivatização pós-coluna, detector fotométrico operando no visível, injetor automático, compartimento de colunas capaz de manter uma temperatura de (35 ± 2)°C e coluna cromatográfica IC Pack Anion HC (150 mm × 4,6 mm) ou similar;
-
Tubos para centrífuga em polipropileno com capacidade para 50 mL;
-
Vial de 2 mL.
¶ Reagentes e soluções
-
Solução ácida de Mo(V)-Mo(VI) – derivatizante:
Dissolver 5,30 g de molibdato de amônio tetrahidratado ((NH₄)₆Mo₇O₂₄·4H₂O, CAS 12027-67-7) em 900 mL de água, com subsequente adição de 100 mL de ácido sulfúrico (H₂SO₄, CAS 7664-93-9) a 98%. Adicionar, sob agitação, 0,65 g de zinco metálico em pó (Zn, CAS 7440-66-6). Após a completa dissolução do zinco, armazenar em frasco âmbar por até 6 meses.
-
Solução de referência de polifosfatos:
Em 900 mL de água Tipo I, adicionar 148 mg de hidrogenofosfato de sódio (Na₂HPO₄, CAS 7558-79-4), 153 mg de pirofosfato de sódio (Na₄P₂O₇, CAS 7722-88-5), 145 mg de trifosfato de sódio (Na₅P₃O₁₀, CAS 7758-29-4) e 129 mg de trimetafosfato de sódio (Na₃P₃O₉, CAS 7785-84-4). Transferir para balão volumétrico de um litro e completar para o volume. Filtrar a solução em membrana de 0,45 μm e armazenar sob refrigeração por até 12 meses. Descartar se observada turbidez ou presença de fungos.
-
Solução tampão borato pH 9,0 – fase móvel:
Em 900 mL de água Tipo I, dissolver 6,18 g (100 mmol) de ácido bórico (H₃BO₃, CAS 10043-35-3), 2,39 g (42,5 mmol) de hidróxido de potássio (KOH, CAS 1310-58-3), 1,49 g (20 mmol) de cloreto de potássio (KCl, CAS 7447-40-7) e 0,484 g (0,13 mmol) de EDTA dissódico (C₁₀H₁₄N₂O₈Na₂·2H₂O, CAS 6381-92-6). Ajustar o pH para 9,0 com auxílio de solução diluída de hidróxido de potássio e/ou solução diluída de ácido bórico, filtrar em membrana de 0,45 μm, transferir para balão volumétrico de um litro e completar para o volume.
Sais de potássio dos ânions polifosfato poderão ser utilizados em substituição aos sais de sódio.
¶ Preparo da amostra
-
Pescado congelado:
Remover, se presente, a água de recobrimento (“glaciamento”) da amostra utilizando água corrente. Retirar porções musculares da amostra ainda congelada e homogeneizar. Analisar imediatamente.
-
Carnes e produtos cárneos:
Retirar porções musculares da amostra ainda congelada e homogeneizar. Analisar imediatamente.
¶ Procedimento de análise
-
Pesar (5,0 ± 0,1) g de amostra em tubo de centrífuga de 50 mL;
-
Adicionar 25 mL de água Tipo I e homogeneizar utilizando homogeneizador tipo Turrax;
-
Deixar em banho-maria acima de 90°C por 5 min, agitando ocasionalmente;
-
Centrifugar por 10 min;
-
Com o auxílio de uma seringa acoplada a filtro de membrana de 0,45 μm, coletar cerca de 1 mL do sobrenadante para o vial de HPLC;
-
Injetar, utilizando os parâmetros constantes da Tabela 6, a solução de referência de polifosfatos e a amostra. A ordem de eluição segue o número de unidades de fosfato: ortofosfato, pirofosfato, trifosfato, trimetafosfato, etc., conforme exemplificado na Figura 3. O volume de injeção deve ser suficiente para que se obtenha uma relação sinal/ruído superior a 3 para cada ânion quando injetada a solução de referência numa diluição 1:15.
| Parâmetro | Valor |
|---|---|
| Detecção | Entre 800 nm e 840 nm |
| Tempo de corrida* | 20 min |
| Temperatura da coluna | (35 ± 2)°C |
| Temperatura do forno de reação pós-coluna | ≥ 115°C |
| Fluxo de derivatizante | 0,2 mL·min⁻¹ |
| Fluxo da fase móvel | 0,5 mL·min⁻¹ |
| Volume de injeção | Entre 1 µL e 75 µL |
retenção superiores a 20 min.
¶ Expressão dos resultados
Considerar “positivo” apenas os picos de polifosfatos integrados no cromatograma com relação sinal/ruído superior a 3.
Reportar “Presença de X (Y) na porção muscular” para cada um dos polifosfatos presentes na solução de referência encontrados, sendo X o nome do ânion polifosfato e Y sua fórmula química. Reportar “Presença de polifosfatos ” se o cromatograma apresentar picos com tempos de retenção superiores ao do trimetafosfato e ausentes da solução de referência. Reportar “não detectado” se o cromatograma apresentar apenas o pico referente ao ânion monofosfato (ortofosfato, ).
¶ Bibliografia
Norimasa Yosa, Izumi Akazaki, Tetsuya Nakazato, Nobuyuki Ueda, Hiroki Kodama e Akira Tateda. High-Performance Liquid Chromatographic Determination of Pyrophosphate in the presence of a 20,000-fold Excess of Orthophosphate. Analytical Biochemistry, v. 199, p. 279 – 285, 1991.
¶ Determinação de conservantes
Utilizar o método descrito nas Seções Produtos cárneos - Ácido benzóico e/ou benzoatos, Produtos cárneos - Ácido sórbico e/ou sorbatos ou Produtos cárneos - Determinação de conservantes por cromatografia líquida acoplada à espectrometria de massas.
¶ Determinação de sulfito por eletroforese capilar de zona
¶ Princípio
Este método se baseia na extração e derivatização do sulfito das amostras de pescado (camarões e lagostas) com acetonitrila e formaldeído por extração sólido-líquido (alternativa 1) ou extração por líquido pressurizado (alternativa 2). A separação do analito na forma de íon hidroximetil sulfonato é realizada por eletroforese capilar de zona e a detecção se dá de forma indireta por arranjo de diodos (CZE-DAD). A quantificação é feita por padronização interna com ácido malônico.
¶ Campo de aplicação
Camarões e lagostas.
¶ Materiais e equipamentos
-
Agitador de tubos;
-
Balança analítica, com resolução mínima de 0,1 mg;
-
Balões volumétricos de 10 mL e 25 mL;
-
Béquer pequeno;
-
Banho-maria com agitação;
-
Banho ultrassônico;
-
Capilares de sílica fundida com revestimento externo de poli-imida com 75 μm de diâmetro interno e 38.5 cm de comprimento total (30 cm de comprimento até o detector);
-
Centrífuga refrigerada;
-
Centrifuga de microtubos;
-
Espátulas de vidro ou silicone;
-
Estantes para tubos tipo “falcon”;
-
Homogeneizador/disruptor de amostras (modelo Ultra Turrax ou similar);
-
Micropipetas e ponteiras para faixas de volume de 1000 μL–10000 μL, 100 μL–1000 μL e 5 μL–200 μL;
-
Microtubos de polipropileno de capacidade mínima de 1,5 mL;
-
Seringas plásticas (capacidade 5 mL ou 10 mL);
-
Sistema automático de eletroforese capilar, com detector de arranjo de diodos, controlador de temperatura, operado por software de controle e aquisição de dados;
-
Sistema de extração por líquido pressurizado, com tubos extratores, filtros de celulose e fibra de vidro;
-
Tubos de polipropileno para extração (tipo “falcon”) de capacidade de 50 mL;
-
Vials para eletroforese capilar.
¶ Reagentes e soluções
-
Acetonitrila (CH₃CN, CAS 75-05-8) de grau cromatográfico;
-
Eletrólito de corrida pH 4,5 (ácido benzenossulfônico 20 mmol·L⁻¹ e ácido aminocaproico 45 mmol·L⁻¹):
Pesar 0,0877 g de ácido benzenossulfônico (C₆H₆O₃S, CAS 98-11-3) e 0,1506 g de ácido aminocaproico (C₆H₁₃NO₂, CAS 60-32-2) em um béquer e diluir em água. Transferir para balão volumétrico de 25 mL e completar volume com água. Filtrar a solução utilizando o sistema de filtração de solvente ou seringa acoplada a membrana de PTFE. Se necessário, ajustar o pH adicionando pequenas massas de um dos dois componentes. Essa solução deve ser preparada no momento do uso.
-
Formaldeído (CH₂O, CAS 50-00-0) 36,5%–38%;
-
Solução de fortificação de íon malonato 120 mg·L⁻¹
-
Solução de fortificação de sulfito derivatizado 10 g·L⁻¹:
Pesar 0,1606 g de sulfito de sódio (Na₂SO₃, CAS 7757-83-7) em um béquer e dissolver em 805 μL de formaldeído. Transferir para balão volumétrico de 10 mL e completar volume com água. Essa solução deve ser preparada no momento de uso.
-
Solução de hidróxido de sódio (NaOH, CAS 1310-73-2) 1 mol·L⁻¹.
-
Solução estoque de íon malonato 1 g·L⁻¹;
-
Solução extratora/derivatizante (0,3% formaldeído e 70% acetonitrila) para extração sólido-líquido (alternativa 1):
Em uma proveta, medir 350 mL de acetonitrila. Acrescentar 4,03 mL de formaldeído. Avolumar com água ultrapura até 500 mL. Essa solução é válida por quatro semanas.
-
Solução extratora/derivatizante (0,3% formaldeído e 40% acetonitrila) para extração líquido pressurizado (alternativa 2):
Em uma proveta, medir 200 mL de acetonitrila. Acrescentar 4,03 mL de formaldeído. Avolumar com água ultrapura até 500 mL. Essa solução é válida por quatro semanas.
-
Terra diatomácea.
¶ Preparo da amostra
-
Crustáceo congelado:
Remover, se presente, a água de recobrimento (“glaciamento”) da amostra utilizando água corrente. Remover a carapaça, cortar porções da musculatura e homogeneizar em moinho ou processador de alimentos. Analisar imediatamente.
-
Crustáceo resfriado:
Remover a carapaça, cortar porções da musculatura e homogeneizar em moinho ou processador de alimentos. Analisar imediatamente.
¶ Procedimento
¶ Alternativa 1
-
Usando balança analítica, pesar (5,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL;
-
Adicionar 10 mL de solução extratora/derivatizante a cada tubo;
-
Homogeneizar o conteúdo do tubo com auxílio de Ultra Turrax;
-
Deixar em banho ultrassônico por 8 min;
-
Aquecer em banho-maria a cerca de 90°C com agitação por 10 min;
-
Centrifugar a no mínimo 3000g por 10 min à temperatura de 4°C;
-
Transferir 500 μL do extrato para um microtubo e adicionar 500 μL de padrão interno (íon malonato 120 mg·L⁻¹) e 500 μL de água;
-
Centrifugar a amostra por 5 min a cerca de 17000g;
-
Transferir cerca de 500 μL do sobrenadante para os vials e injetar no sistema de eletroforese.
-
Preparar uma curva de calibração em matriz. Pesar (5,0 ± 0,1) g de amostra branca em tubos de polipropileno de 50 mL e fortificar com sulfito derivatizado na faixa de trabalho de 25 mg·L⁻¹ a 200 mg·L⁻¹. Preparar também uma amostra de recuperação fortificada no limite regulatório. Proceder à extração conforme a descrição acima, diluir com padrão interno e injetar no sistema de eletroforese.
¶ Alternativa 2
-
Usando balança analítica, pesar (5,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL;
-
Adicionar aproximadamente 3 g de terra diatomácea nos tubos contendo as amostras e homogeneizar até completa dispersão com auxílio de um bastão de vidro;
-
Transferir a amostra para o tubo extrator com filtro;
-
Extrair as amostras em sistema de extração por líquido pressurizado utilizando 30 mL de solução extratora com 0,3% formaldeído e 40% de acetonitrila por 5 min a 90°C;
-
Após extração, resfriar os tubos com os extratos em freezer por 30 min a −18°C;
-
Centrifugar a no mínimo 3000g por 10 min à temperatura de 4°C;
-
Transferir 960 μL do extrato para um microtubo e adicionar 40 μL de padrão interno (íon malonato 1 g·L⁻¹);
-
Centrifugar a amostra por 5 min a cerca de 17000 g;
-
Transferir cerca de 500 μL do sobrenadante para os vials e injetar no sistema de eletroforese.
-
Preparar uma curva de calibração em matriz. Pesar (5,0 ± 0,1) g de amostra branca em tubos de polipropileno de 50 mL e fortificar com sulfito derivatizado na faixa de trabalho de 25 mg·L⁻¹ a 200 mg·L⁻¹. Preparar também uma amostra de recuperação fortificada no limite regulatório. Proceder à extração conforme a descrição acima, diluir com padrão interno e injetar no sistema de eletroforese.
¶ Parâmetros instrumentais recomendados para separação
-
Injeção hidrodinâmica: 50 mbar ⁻¹ 5s;
-
Comprimento de onda: 210 nm;
-
Tensão de separação: −30 kV;
-
Temperatura do capilar: 20°C;
-
Condicionamento do capilar: 1 mol·L⁻¹ – 15 min; água – 15 min; eletrólito – 15 min;
-
Pré-condicionamento entre corridas: eletrólito – 1 min.
¶ Expressão dos resultados
A identificação do analito é feita pelo seu tempo de migração e a quantificação é realizada por padronização interna (50 mg·L⁻¹), utilizando relação funcional linear de razão das concentrações (eixo x) versus razão de áreas dos picos (eixo y). Expressar o resultado da amostra em “g de /100 g”, com três casas decimais utilizando-se a seguinte equação:
¶ Bibliografia
Samantha Gonçalves, Luciano Molognoni, Heitor Daguer, Ricardo Pimenta, Rodrigo Barcellos Hoff, Gustavo Amadeu Micke e Luciano Vitali. Simultaneous extraction/derivatization for the analysis of sulfite by capillary electrophoresis: A high-throughput reference method to meet the demand of seafood inspection. Food Research International, vol. 161, p. 111780, 2022.
¶ Determinação do teor de fósforo total
Determinar o teor de fósforo total utilizando um dos métodos descritos na norma ISO 23776 ou AOAC 2011.14, reportando o valor obtido em "g·kg⁻¹" com duas casas decimais. Se necessário expressar o resultado em P₂O₅, utilizar 2,29 como fator de conversão de P para P₂O₅.
Amostras “glaciadas” devem ter a água de recobrimento removida com auxílio de água corrente antes do ensaio, tomando-se a precaução de não descongelar substancialmente a amostra.
¶ Histamina
Determinar o teor de histamina em uma amostra composta por nove unidades amostrais utilizando o método descrito na norma NMKL 196 ou na Seção Pescados e subprodutos da pesca - Histamina por cromatografia líquida acoplada à espectrometria de massas, reportando o valor de cada unidade amostral e sua média em “mg/kg” como um número inteiro.
Obter uma nova curva de calibração a cada batelada.
¶ Histamina por cromatografia líquida acoplada à espectrometria de massas
¶ Princípio
O método se baseia na extração sólido-líquido com partição em baixa temperatura, utilizando mistura de solventes orgânicos com baixa proporção de água, acidificados com ácido acético. A separação dos analitos é realizada através da cromatografia líquida em fase reversa, utilizando fase estacionária de cianopropil. A quantificação é feita por espectrometria de massas em modo tandem, utilizando fonte de ionização por electrospray (LC-ESI-MS/MS) e padronização interna.
¶ Campo de aplicação
Pescado e produtos derivados.
¶ Materiais e equipamentos
-
Agitador de tubos tipo vortex;
-
Agitador orbital;
-
Balança analítica;
-
Balões volumétricos de 10 mL;
-
Banho de ultrassom;
-
Centrífuga refrigerada para tubos de 50 mL;
-
Centrífuga refrigerada para microtubos;
-
Coluna para cromatografia líquida com fase estacionária de di-isopropril-3-cianopropil silano ligado à sílica hidroxilada, com partículas de diâmetro nominal mínimo de 3,5 μm (Zorbax-Agilent SB-CN, ou similar);
-
Dispensador automático de 50 mL;
-
Homogeneizador/disruptor de amostras (modelo Ultra Turrax ou similar);
-
Micropipetadores e ponteiras para volumes de 2-20 μL, 10-100 μL, 100-1000 μL e 1000-5000 μL;
-
Microtubos de polipropileno, com capacidade para 1,5 mL;
-
Provetas de 250 mL e 500 mL;
-
Sistema de cromatografia líquida acoplada a espectrômetro de massas em modo tandem, com fonte de ionização por electrospray (LC-ESI-MS/MS);
-
Tubos de polipropileno, com capacidade para 15 mL e 50 mL (tipo falcon);
-
Vials para HPLC de borossilicato com capacidade de 1,5 mL.
¶ Reagentes e soluções
-
Solução extratora de acetonitrila (CH₃CN, CAS 75-05-8) : metanol (CH₃OH, CAS 67-56-1) : água (45:45:10 v/v/v) acidificada com 0,1% ácido acético (CH3COOH, CAS 64-19-7).
-
Fase móvel A (solução aquosa de ácido fórmico (HCOOH, CAS 64-18-6) 0,1%);
-
Fase móvel B (solução de acetonitrila acidificada com 0,1% de ácido fórmico);
-
Fase móvel de diluição (fase móvel A:fase móvel B 90:10 v/v);
-
Soluções-estoque de histamina (C₅H₉N₃, CAS 51-45-6) 10 g·L⁻¹, preparada em metanol;
-
Solução-estoque do padrão interno 1,3-diaminopropano (C₃H₁₀N₂, CAS 109-76-2) 10 g·L⁻¹, preparada em metanol com baixa proporção de água para dissolução;
-
Solução de fortificação de histamina 1 g·L⁻¹, preparada em metanol, a partir da solução-estoque.
-
Solução de fortificação do padrão interno 1,3-diamino-propano 100 mg·L⁻¹, preparada em metanol, a partir da solução-estoque;
Obs.: os solventes orgânicos devem ser de grau LC-MS e a água deve ser de grau ultrapuro.
¶ Preparo da amostra
-
Pescado in natura e salgado:
Limpar o pescado retirando a pele e escamas, espinha, gordura e vísceras. Retirar porções da musculatura de várias regiões do pescado e cortar as amostras em pedaços menores para processar e homogeneizar em processador.
-
Conservas de pescado:
Processar e homogeneizar todo o conteúdo da lata (líquido + parte sólida) em processador.
¶ Procedimento de análise
-
Utilizando balança analítica, pesar (2,0 ± 0,1) g de amostra em tubos de polipropileno de capacidade 50 mL;
-
Fortificar as amostras com 200 μL de padrão interno;
-
Adicionar 10 mL de solução de extração aos tubos com amostra;
-
Homogeneizar o conteúdo de cada tubo por volta de cinco segundos no homogenizador/disruptor;
-
Agitar em mesa agitadora orbital durante 20 min;
-
Centrifugar por 10 min à rotação de no mínimo 3000g, à temperatura de 4°C;
-
Transferir o sobrenadante para um tubo de polipropileno de 15 mL de capacidade e levar ao freezer (à temperatura de cerca de −18°C) por no mínimo uma hora;
-
Centrifugar novamente por 10 min à rotação de no mínimo 3000g, à temperatura de 4°C;
-
Transferir 50 μL do sobrenadante do extrato para vials e completar com 950 μL de fase móvel de diluição (90A:10B). Homogeneizar e injetar no sistema LC-ESI-MS/MS. Caso necessário, ajustar essa diluição à sensibilidade do equipamento.
-
Preparar uma curva de calibração em matriz, pesando-se (2,0 ± 0,1) g de amostra “branca” para cada ponto e fortificando com histamina (25 mg·L⁻¹ a 200 mg·L⁻¹) e o padrão interno (100 mg·L⁻¹). Preparar também uma amostra de recuperação em triplicata, fortificada no limite regulatório. Seguir os procedimentos de extração acima.
-
Parâmetros instrumentais: temperatura do forno de coluna: 40°C; volume de injeção: 10 μL; fluxo da fase móvel: 0,5 mL min⁻¹; gradiente de eluição: 0-1 min, 90% A; 2-3 min, 80% A; 4 min, 70% A, 5-6 min, 50% A; 7-8 min, 10% A; 9 min, 50% A; 10 min, 90% A; mais 4 min para auto-equilíbrio do sistema;
-
Parâmetros de espectrometria de massas: íons precursores: m/z 112,1 (histamina) e m/z 75,1 (1,3-Diaminopropano); transições de quantificação: m/z 54,1 (histamina) e m/z 58.0 (1,3-Diaminopropano); transição de confirmação: m/z 68,1 (histamina).
¶ Cálculo e expressão dos resultados
Utilizando o método dos mínimos quadrados, obter, a partir dos cromatogramas dos padrões, uma curva de calibração usando a concentração de histamina em mg·kg⁻¹ versus a razão de áreas dos picos cromatográficos (histamina/padrão interno). Aceitar a curva para valores de R² maiores que 0,95. Identificar no cromatograma da amostra o pico com o mesmo tempo de retenção dos padrões de histamina e razão iônica superior a 20% (transição de quantificação/transição de confirmação). Calcular a concentração do analito na amostra utilizando a equação da curva de calibração. Expressar o resultado da amostra em “mg/kg” como um número inteiro.
¶ Bibliografia
Luciano Molognoni, Heitor Daguer, Leandro Antunes de Sá Ploêncio, Juliano de Dea Lindner. A multi-purpose tool for food inspection: simultaneous determination of various classes of preservatives and biogenic amines in meat and fish products by LC-MS. Talanta, vol. 178, p. 1053-1066, 2018.
¶ Índice de peróxidos
Utilizar o método descrito na norma ISO 3960 para determinação do Índice de Peróxidos na amostra, em “meq de O₂/kg”. Se necessário, corrigir o valor obtido pelo teor de gordura, expressando o resultado final em “meq de O₂/kg de gordura” com uma casa decimal.
¶ Lipídios totais
Determinar o teor de Lipídios Totais utilizando o método descrito na norma ISO 1443 ou, opcionalmente, na norma NMKL 181 ou na norma AOAC 2008.06. Reportar o valor obtido em “g/100 g” com uma casa decimal.
¶ Nitritos e Nitratos
Determinar, utilizando o método descrito nas normas NMKL 165 (nitritos e nitratos), NMKL 194 (nitritos e nitratos) ou ISO 2918 (nitritos) e ISO 3091 (nitratos), o teor de nitritos e nitratos na amostra, expressando os resultados obtidos em “mg de /kg” como um número inteiro. Alternativamente, utilizar o método descrito na Seção Produtos cárneos - Nitritos e nitratos por eletroforese capilar de zona.
¶ pH
Determinar o pH no extrato homogeneizado da amostra, conforme metodologia descrita na norma ISO 2917. Reportar o valor obtido com duas casas decimais, arredondando para 0,05 unidades de pH.
¶ Potássio
Determinar o teor de potássio na amostra utilizando o método descrito na norma AOAC 969.23 ou AOAC 2011.14, reportando o resultado obtido em “mg de K/100 g” como um número inteiro.
Amostras “glaciadas” devem ter a água de recobrimento removida com auxílio de água corrente antes do ensaio, tomando-se a precaução de não descongelar substancialmente a amostra.
Obter uma nova curva de calibração a cada batelada.
¶ Proteína
Determinar o teor de nitrogênio na amostra de acordo com a norma ISO 1871. Multiplicar o valor obtido por 6,25 para obtenção do teor de proteína. Expressar o resultado obtido em “g/100 g” com duas casas decimais. Alternativamente, utilizar a norma AOAC 992.15.
Amostras “glaciadas” devem ter a água de recobrimento removida com auxílio de água corrente antes do ensaio, tomando-se a precaução de não descongelar substancialmente a amostra.
¶ Relação Umidade/Proteína
A partir dos resultados para o teor de umidade e teor de proteínas, obtidos utilizando-se os métodos estabelecidos neste Manual, calcular a relação U/P, expressando o resultado final com duas casas decimais.
¶ Resíduo mineral fixo (Cinzas)
Determinar o teor de resíduo mineral fixo (Cinzas) utilizando o método descrito na norma ISO 936, reportando o resultado obtido em “g/100 g” com uma casa decimal.
¶ Sódio
Utilizando o método descrito na norma AOAC 969.23 ou AOAC 2011.14, determinar o teor de sódio na amostra. Reportar o resultado obtido em “mg de Na/100 g” como um número inteiro.
Amostras “glaciadas” devem ter a água de recobrimento removida com auxílio de água corrente antes do ensaio, tomando-se a precaução de não descongelar substancialmente a amostra.
Obter uma nova curva de calibração a cada batelada.
¶ Umidade
Determinar o teor de Umidade utilizando o método descrito na norma ISO 1442, reportando o resultado obtido em “g/100 g” com uma casa decimal. Alternativamente, utilizar o método descrito na norma AOAC 2008.06.
Amostras “glaciadas” devem ter a água de recobrimento removida com auxílio de água corrente antes do ensaio, tomando-se a precaução de não descongelar substancialmente a amostra.
¶ Peixe salgado e salgado seco
Determinar o teor de Umidade utilizando o método descrito na norma AOAC 950.46B item (a), reportando o resultado obtido em “g/100 g” com uma casa decimal.
Bacalhau espalmado e similares (p. ex. Saithe) devem ser amostrados e analisados de acordo com o método descrito na seção Umidade (método cross section).
¶ Umidade (método cross section)
¶ Princípio
O peixe é cortado em seções conforme descrito no método. As seções são cortadas em pedaços menores para formar a amostra. A umidade da amostra é determinada por secagem em estufa à temperatura de 103°C a 105°C.
¶ Campo de aplicação
Este método se aplica a amostras de peixe salgado e de peixe salgado seco da família Gadidae que foi totalmente saturado com sal ou a amostras de peixe salgado que foi conservado por saturação parcial em sal de teor não inferior a 12%.
¶ Materiais e equipamentos
- Balança com resolução de 1 g;
- Dessecador;
- Estufa de secagem;
- Faca;
- Pincel (ou escova);
- Recipientes para secagem (aço, vidro ou porcelana);
- Serra de fita;
- Tesoura.
¶ Preparo da amostra
- Remover cristais de sal superficiais com pincel (ou escova), sem água.
- Registrar massa do peixe (resolução 1 g).
- Medir o comprimento do peixe: distância entre a fenda da cauda e a linha traçada entre as pontas dos ossos da orelha.
Obs.: Pode ser difícil cortar seções de 2 mm quando o peixe tem um teor de água superior a 50%, mas a seção deve ser próxima de 2 mm.
Para minimizar a perda de água das seções de 2 mm é importante pesar a amostra coletada imediatamente após o peixe ser cortado em seções.
As amostras devem ser armazenadas sob refrigeração ou refrigeradas desde o momento da amostragem (corte em seções) até o momento da análise.
A análise deve ser realizada o mais rápido possível após o corte em seções.
¶ Procedimento de análise
A. Tara dos recipientes de secagem
- Acondicionar os recipientes que serão utilizadas no ensaio em estufa à temperatura entre 103°C e 105°C por uma hora.
- Resfriar em dessecador e pesar.
B. Corte e coleta (cross section)
A amostragem dos peixes deve ser feita conforme a descrição da Figura 4. Cortar as amostras em seções, com faca ou serra de fita. O peixe é cortado em seções de 40 mm, das quais são cortadas seções de 2 mm, que são utilizadas para formar a amostra na qual será determinada a umidade, procedendo-se como se segue:
- Cortar uma seção de 20 mm, medida a partir de uma linha traçada entre os ossos da orelha (linha pontilhada da figura);
- Em seguida, cortar uma seção de 40 mm;
- Cortar outra seção de 2 mm da parte frontal da seção de 40 mm e coletar;
- Cortar novamente uma seção de 40 mm;
- Cortar outra seção de 2 mm da parte frontal da seção de 40 mm e coletar;
- Colher as seções de 2 mm de números pares (conforme marcações II, IV, VI, VIII da Figura 4) para formar a amostra;
- Cortar as seções de 2 mm com tesoura, obtendo pedaços menores que serão depositados diretamente em recipientes de secagem logo após esse procedimento;
- Pesar os recipientes que contêm a amostra.
C. Secagem
- Acondicionar os recipientes contendo as amostras em estufa à temperatura de 103°C a 105°C para secagem até peso constante, por no mínimo 18 horas.
- Acondicionar os recipientes em dessecador para seu resfriamento e pesar em seguida.
¶ Cálculo e expressão dos resultados
Determinar a umidade conforme a equação:
onde W1 = massa dos peixes e recipientes antes da secagem (em g); W2 = massa dos peixes e recipientes após secagem (em g); Ws = peso das recipientes taradas (em g).
Obs.: O resultado é reportado até o grama mais próximo.
¶ Bibliografia
Codex Alimentarius. Codex Standard for Salted Fish and Dried Salted Fish of the Gadidae Family of Fishes – Codex Stan 167-1989. Roma: FAO/WHO, 2016.
¶ Verificação das formas de apresentação de conservas de pescado e meio de cobertura
¶ Princípio
Este método se baseia na separação física e pesagem, em condições controladas, do conteúdo interno de conservas de pescado para determinação do peso líquido drenado, percentual de carne em relação ao peso líquido drenado, percentual de água em relação ao peso líquido declarado, percentual de óleo, percentual de pedaços soltos em relação ao peso líquido declarado e percentual de meio de cobertura em relação ao peso líquido declarado.
¶ Campo de aplicação
Conservas de pescado com até 300 g de peso líquido declarado.
¶ Materiais e equipamentos
-
Abridor de latas;
-
Aparato para separação do meio de cobertura da carne efetiva de conservas de pescado, composto por peneiras com malha 2,8 mm x 2,8 mm em aço inoxidável dispostas em ângulo de 17° a 20° para drenagem de líquidos com massa especifica entre 0,80 a 1,1 g·mL⁻¹ e com volume morto inferior a 1% do líquido drenado;
-
Balança, com resolução mínima de 0,1 g;
-
Cronômetro;
-
Espátulas;
-
Peneira com malha de 12 mm x 12 mm em aço inoxidável com bandeja de apoio;
-
Pinças;
-
Provetas com capacidade de 100 mL, resolução mínima de 1 mL.
¶ Reagentes e soluções
- Etanol (C₂H₅OH, CAS 64-17-5).
¶ Preparo da amostra
Cada item de ensaio é composto por no mínimo seis unidades em suas embalagens originais e mantido à temperatura ambiente. Antes de se iniciar a execução deste método analítico, o item de ensaio deve ser mantido à temperatura entre 20ºC e 30°C por no mínimo 12 h.
¶ Procedimento de análise
-
Limpar os recipientes das conservas utilizando papel toalha e álcool;
-
Numerar os recipientes sequencialmente;
-
Registrar as informações declaradas no rótulo do item de ensaio: peso líquido drenado declarado (PDd, em g) e peso líquido declarado (PLq, em g),
-
Pesar a proveta e registrar o valor PPr (massa da proveta, em g);
-
Pesar a peneira 1 (2,8 mm x 2,8 mm) e registrar o valor PP (massa da peneira 1, em g);
-
Abrir os recipientes tomando cuidado para não perder frações do meio de cobertura durante o processo;
-
Verter cada recipiente na peneira 1, espalhar todo o conteúdo sobre a malha com auxilio de espatulas e drenar por 2 min, usando cronômetro;
-
Recolher o meio de cobertura diretamente na proveta previamente pesada;
-
Pesar novamente peneira 1 com o conteúdo drenado e registrar o valor PPP (massa da peneira 1 + carne, em g);
Observação: para latas contendo molhos e/ou ingredientes opcionais como condimentos, vegetais e frutas que sejam visíveis, o conteúdo deve ser lavado na peneira 1 antes da pesagem. Para isso, lavar toda a carne com água quente (≈ 40°C) até eliminar o molho residual aderido. Se necessário, separar os ingredientes opcionais visíveis, utilizando pinça;
-
Pesar a proveta + meio de cobertura drenado e registrar o valor PPL (massa da proveta + líquido de cobertura, em g);
-
Aferir o volume total do meio de cobertura na proveta e registrar o valor VT (em mL);
-
Aferir o volume da fase aquosa recolhida na proveta e registrar o valor VFA (em mL). Utilizar valores de massa específica (ρ, em g·mL⁻¹) correspondente às frações de líquidos declaradas pelo fabricante para estimar os percentuais mássicos (Tabela 7). Utilizar valores para ρ até a obtenção de erro relativo inferir ou igual a 5% em relação à massa total do meio de cobertura;
-
Pesar a base da peneira 2 (12 mm x 12 mm) e registrar o valor R (massa da base da peneira 2, em g);
-
Transferir todo o conteúdo da peneira 1 para a peneira 2, com esta última apoiada à base previamente tarada, coletando os pedaços de peixe que passaram pela malha (12 mm x 12 mm);
-
Espalhar todo o conteúdo utilizando espátula com cuidado para não desconfigurar as peças. Remover os pequenos pedaços que aderiram à tela e agregar à pesagem;
-
Pesar a base da peneira 2 + pedaços de peixe e registrar o valor RP (massa da base da peneira 2 + pedaços soltos de peixe, em g).
| Meio de cobertura | faixa de ρ, em g·mL⁻¹ |
|---|---|
| Óleo de soja | 0,91 a 0,92 |
| Azeite de oliva | 0,90 a 0,91 |
| Molho de tomate; caldo vegetal | 1,04 a 1,07 |
¶ Cálculo e expressão dos resultados
Determinar, para cada unidade:
- O peso drenado (PD, em g):
- O percentual de carne:
- O percentual de água no meio de cobertura:
- O percentual de óleo no meio de cobertura:
- O percentual de pedaços soltos:
- O percentual de meio de cobertura:
- O erro na estimativa da massa específica ρ das frações de meio de cobertura:
em que ρfo é a massa específica da fase oleosa, em g·mL⁻¹ e ρfa é a massa específica da fase aquosa, em g·mL⁻¹.
Reportar cada conjunto de resultados por lata, conforme as unidades e as regras descritas abaixo:
-
Percentual de água em relação ao peso líquido declarado, sem casas decimais;
-
Percentual de carne em relação ao peso líquido declarado, sem casas decimais;
-
Percentual de meio de cobertura em relação ao peso líquido declarado, sem casas decimais;
-
Percentual de óleo em relação ao peso líquido declarado, sem casas decimais;
-
Percentual de pedaços soltos em relação ao peso líquido drenado declarado, sem casas decimais;
-
Peso líquido drenado (em g), sem casas decimais;
-
Adicionalmente, informar no campo “Observações” do certificado o peso líquido e o peso líquido drenado declarados pelo fabricante.
¶ Bibliografia
Codex Alimentarius. Codex Standard for canned tuna and bonito – CODEX STAN 70 rev. 1. Roma: FAO/WHO, 1995.
¶ Disposições Gerais
As sugestões para aprimoramento ou possíveis correções deste documento devem ser direcionadas ao Departamento responsável, para alinhamento das melhores práticas de mercado, legislação vigente e/ou regulamentações, que não tenham sido contempladas na versão vigente.
¶ Histórico de revisões
- 8ª ed., 9 de março de 2026
-- Descrição do procedimento do método de umidade cross section (item 5.25) e atualização da numeração dos métodos seguintes. Doc. SEI n° 50875716.
-- Inclusão do método de detecção de β-lactoglobulina em queijos (item 2.11) e atualização da numeração dos métodos seguintes. Doc. SEI n° 50875716. - 7ª ed., 25 de fevereiro de 2026
-- Alteração do preparo de amostras de produtos cárneos fatiados destinadas à análise de conservantes (itens 1.1, 1.2, 1.3 e 1.17). Doc. SEI n° 50670510.
-- Atualizada a equipe técnica (inclusão de Renan Maciel). - 6ª ed., 6 de fevereiro de 2026
-- Reinclusão da norma IDF 139 para análise de sorbatos em produto lácteos. Doc. SEI nº 50259487.
-- Inclusão de Carolina Maso Viegas à equipe técnica em substituição a Tiago Souza da Cunha. - 5ª ed., 8 de agosto de 2025
-- Inclusão do método IDF 20-1 para o ensaio de proteína em produtos lácteos. - 4ª ed., 8 de agosto de 2025
-- Inclusão do método AOAC 2011.14 para Ca, P, Na e K. Alteração no método para formaldeído em leite, alteração no escopo da determinação de açúcares por cromatografia iônica, remoção do método IDF 139 em favor da NMKL 124. Doc. SEI n° 44686031. - 3ª ed., 13 de agosto de 2024
-- Alteração na expressão do resultado do ensaio atividade de água. - 2ª Ed., 30 de julho de 2024
-- Publicação em formato wiki, disponível no repositório de manuais da SDA, conforme preconizado pela Portaria SDA/MAPA nº 1110, 13 de maio de 2024.
-- Alterada a unidade de concentração de aditivos alimentares, como os conservantes, cujos resultados passam a ser expressos em mg·kg⁻¹, em conformidade com as novas unidades adotadas pela Anvisa em seus regulamentos.
-- Incluído o método de referência da ISO para análise de corantes artificiais em produtos cárneos.
-- Atualizada a numeração de diversos métodos de referência da IDF para análise de leite e produtos derivados.
-- Acrescentado um novo método analítico que contempla o controle de conservas de pescado.
-- Extensão de escopo para o método de detecção de gordura vegetal em novas matrizes lácteas.
-- Substituição do método NMKL 148 por método descrito neste manual para a análise de sacarose e lactose em leite e produtos lácteos