¶ Folha de rosto
© 2025 Ministério da Agricultura e Pecuária. Todos os direitos reservados. É permitida a reprodução parcial ou total desta obra, desde que citada a fonte e que não seja para venda ou qualquer fim comercial. A responsabilidade pelos direitos autorais de textos e imagens desta obra é do autor.
Ano 2025
Elaboração, distribuição, informações:
Ministério da Agricultura e Pecuária
Secretaria de Defesa Agropecuária - SDA
Departamento de Serviços Técnicos - DTEC
Esplanada dos Ministérios, Bloco D, Anexo, Ala B, 4º andar, sala 433
CEP: 70043-900, Brasília - DF
www.agricultura.gov.br
e- mail: cgal@agro.gov.br
Central de Relacionamento: 0800 704 1995
Equipe Técnica:
Coordenação Geral:
Arilson Lehmkuhl (CDL/CGAL)
Erick Soares Lins (CDI/CGAL)
Coordenação Técnico-Científica:
Nélio Fleury Filho (LFDA/GO)
Redação e Desenvolvimento:
Carlos Juliano da Silva (LFDA/SP)
Daiane Cioato (LFDA/RS)
Daniel Rodrigo Hillesheim (LFDA/RS)
Gabriel Barros de Oliveira (LFDA/MG)
Jose Ailton Gonçalves (LFDA/MG)
Leonardo Francisco de Souza (LFDA/MG)
Nélio Fleury Filho (LFDA/GO)
Rafael Pissinatti (LFDA/MG)
Renan de Almeida Maciel (LFDA/PA)
Rodrigo Barcellos Hoff (SLAV/SC)
Vanessa Goncalves dos Santos (LFDA/SP)
¶ Folha resumo
|
Macroprocesso: Laboratórios |
Objetivo: Realizar a padronização, harmonização, atualização e a unificação dos procedimentos para execução e garantia da qualidade de ensaios laboratoriais da área de Resíduos e Contaminante em Alimentos |
|||
|
Processo: Análises Laboratoriais |
||||
|
Entrega: Segurança e qualidade de alimentos |
Público alvo e demais interessados: Laboratórios oficiais ou credenciados do Ministério da Agricultura e Pecuária (Mapa) |
Versão do documento: 2 |
||
|
Setor responsável e responsabilidades A Coordenação Geral de Laboratórios Agropecuários do Departamento de Serviços Técnicos é responsável pela elaboração, atualização e envio para aprovação deste manual, tendo responsabilidade quanto aos procedimentos descritos no documento. |
||||
¶ 1. Definições e Conceitos
¶ 1.1. Definições
Acurácia ou Exatidão: Grau de concordância entre um valor medido e um valor verdadeiro de um mensurando.
Grau de concordância entre o resultado de um ensaio e o valor de referência aceito. A acurácia é determinada através da veracidade e da precisão.
Amostra Branca: Matriz isenta da substância a analisar, ou cujo nível de concentração seja suficientemente baixo de modo a não interferir nos resultados de medição.
Amostra de Laboratório ou Amostra Laboratorial: Amostra enviada para e recebida pelo laboratório.
Amostras de Controle de Qualidade – ACQ: São amostras de matrizes brancas adicionadas, ou não, com o analito. As ACQ são usadas para monitorar o desempenho de análises químicas e para avaliar a integridade, validade e confiabilidade dos resultados.
Amostra de Ensaio ou Amostra Analítica: Amostra preparada a partir da amostra de laboratório, da qual são retiradas as “alíquotas ou porções de ensaio” ou “alíquotas ou porções analíticas”. A amostra laboratorial depois da remoção de quaisquer partes que não serão analisadas, e.g. ossos, solo aderido.
Amostra Fortificada: Amostra branca na qual se adiciona (fortifica) o analito em uma concentração determinada. Tais amostras podem ser utilizadas na validação do método, na rotina analítica como ACQ, ou mesmo para preparo da curva de calibração de matriz fortificada (CCF).
Analista: Responsável por realizar a análise de uma amostra.
Analito: A espécie química, substância, que será identificada e/ou cuja concentração (ou massa) será determinada em uma amostra de ensaio.
Bias: ver “Viés”.
Batelada (de análise): Sinônimo de Lote de análise. Para processos similares de extração e purificação uma batelada de análise é uma série de amostras tratadas por um analista (ou equipe de analistas) em paralelo, geralmente em um dia.
Branco de Reagentes: Uma análise completa realizada utilizando apenas solventes e reagentes, na ausência de qualquer amostra (a amostra pode ser substituída por água, para tornar a análise realista).
Capacidade de Detecção para Triagem – CCβ: É o teor mais baixo que pode ser detectado ou quantificado numa amostra com uma probabilidade de erro β.
Chemical Abstracts Service – CAS: Serviço de Resumos Químicos.
Concentração Retrocalculada (xc): Ao avaliar linearidade, a concentração retrocalculada é o valor de x calculado/predito (xc), obtida pelo sinal (yi) dos pontos da curva de calibração.
Condição de Precisão Interna: Condição de medição num conjunto de condições, as quais compreendem o mesmo procedimento de medição, o mesmo local e medições repetidas no mesmo objeto ou em objetos similares, ao longo de um período extenso, mas pode incluir outras condições que envolvam mudanças.
Condição de Repetitividade: Condição de medição num conjunto de condições, as quais compreendem o mesmo procedimento de medição, os mesmos operadores, o mesmo sistema de medição, as mesmas condições de operação e o mesmo local, assim como medições repetidas no mesmo objeto ou em objetos similares durante um curto período.
Condição de Reprodutibilidade: Condição de medição num conjunto de condições, as quais compreendem diferentes locais, diferentes operadores, diferentes sistemas de medição e medições repetidas no mesmo objeto ou em objetos similares.
Controle de Qualidade – CQ: Conjunto de atividades do sistema de gestão da qualidade focado em demonstrar que os requisitos de qualidade são atendidos. É constituído do conjunto de atividades planejadas para monitorar, verificar e controlar a qualidade dos resultados analíticos, tais como: a análise de amostra branca, ou de amostra de controle, ou de material de referência certificado, a elaboração de cartas de controle e a participação em avaliações externas de qualidade, como os ensaios interlaboratoriais colaborativos e de proficiência.
Controle de Qualidade Interno – CQI: conjunto de ações que um laboratório de química analítica pode utilizar para assegurar que os resultados produzidos estão adequados à finalidade proposta. Na prática, a adequação ao propósito é determinada por uma comparação entre a exatidão obtida no laboratório em um tempo determinado com o nível de exatidão requerido. O CQI, portanto, inclui procedimentos práticos de rotina que possibilitam ao analista aceitar um resultado ou grupos de resultados como adequados ao propósito ou rejeitar os resultados e repetir as análises.
Curva de Calibração: Expressão da relação entre uma indicação e o valor medido correspondente.
Curva de Calibração em Solvente (CCS): Curva de Calibração na qual os seus pontos de calibração são preparados a partir de adição de uma solução padrão em um solvente (ou reagente) usual, normalmente esse solvente deve ser o mesmo presente no final das etapas de extração/digestão e purificação do método.
Curva de Calibração em Matriz Fortificada (CCF): Curva de Calibração na qual os seus pontos de calibração são preparados a partir de adição de uma solução padrão em uma amostra branca antes das etapas de extração/digestão. Os pontos são então analisados instrumentalmente após todas as etapas de extração/digestão e purificação. Este tipo de curva de calibração é o que mais simula as condições reais que amostra sofre durante o processo analítico, por esse motivo o uso de tal curva é indicado para minimizar possíveis efeitos matriz e compensar por erros originários da recuperação do analito.
Curva de Calibração Matrizada (CCM): Curva de Calibração na qual os seus pontos de calibração são preparados a partir de adição de uma solução padrão em um extrato/digerido de uma amostra branca, após as etapas de extração/digestão e purificação. O preparo de tal curva visa avaliar possíveis efeitos matriz e o seu uso em rotina é recomendado para minimizar esses efeitos.
Desvio Padrão de Repetitividade (sr): Desvio-padrão amostral (experimental) calculado de resultados gerados sobre condições de repetitividade.
Desvio Padrão de Reprodutibilidade (sR): Desvio-padrão amostral (experimental) calculado de resultados gerados sobre condições de reprodutibilidade.
Desvio Padrão Relativo de Repetitividade – DPRr, Repeatability Relative Standard Deviation – RSDr, ou Coeficiente de Variação de Repetitividade – CVr: Desvio-padrão relativo calculado de resultados gerados sobre condições de repetitividade:

Desvio Padrão Relativo de Reprodutibilidade – DPRR, Reproducibility Relative Standard Deviation – RSDR ou Coeficiente de Variação de Reprodutibilidade – CVR: Desvio-padrão relativo calculado de resultados gerados sobre condições de reprodutibilidade:

Efeito Matriz: Uma influência de um ou mais compostos co-extraídos da amostra na medição da concentração ou massa do analito. Pode ser observado como um ganho ou perda da resposta do detector em comparação com aquela produzida pelas soluções dos analitos em solventes (livre de componentes da matriz). A presença ou ausência de tais efeitos podem ser demonstradas pela diferença de resposta do padrão no extrato da matriz e do padrão no solvente.
Efeito Matriz Relativo: Diferença na resposta analítica entre um padrão em solvente e um padrão em extrato de matriz (ex. Um ponto da Curva de Calibração Matrizada - CCM) com correção por um padrão interno.
Erro Aleatório: Componente do erro de medição que, em medições repetidas, varia de maneira imprevisível.
Erro Alfa (α): A probabilidade de que a amostra testada seja conforme, mesmo que um resultado não conforme tenha sido obtido.
Erro Beta (β): A probabilidade de que a amostra testada seja verdadeiramente não conforme, mesmo que um resultado conforme tenha sido obtido.
Erro Sistemático: Componente do erro de medição que, em medições repetidas, permanece constante ou varia de maneira previsível.
Exatidão ou Acurácia: ver “Acurácia”.
Faixa de Trabalho – FT ou Faixa de Medição ou Faixa validada: Conjunto de valores de um mensurando para os quais os erros do procedimento medição analítica estão dentro de limites especificados.
Falso Negativo: Um resultado que indica erroneamente que a concentração do analito não excede um valor especificado.
Falso Positivo: Um resultado que indica erroneamente que a concentração do analito excede um valor especificado.
Fator de Abrangência: Número maior do que um pelo qual uma incerteza padrão combinada é multiplicada para se obter uma incerteza de medição expandida.
Heteroscedasticidade: Antônimo de homoscedasticidade.
Homoscedasticidade: Igualdade dos desvios-padrão de diferentes amostras estatísticas. Em uma curva de calibração refere-se à igualdade estatística dos desvios-padrão das replicatas das respostas instrumentais em diferentes níveis de concentração das soluções padrão de calibração.
HorRatR – Reproducibility Horwitz Ratio, Razão de Horwitz de Reprodutibilidade: O desvio-padrão relativo de reprodutibilidade – DPRR ou coeficiente de variação de reprodutibilidade – CVR, dividido pelo desvio-padrão relativo de reprodutibilidade – DPRRH ou coeficiente de variação de reprodutibilidade – CVRH estimado através da equação de Horwitz, na qual w é a fração de massa do analito (com ambos, numerador e denominador, nas mesmas unidades):
HorRatr – Repeatability Horwitz Ratio, Razão de Horwitz de Repetitividade: O desvio-padrão relativo de repetitividade – DPRr, ou coeficiente de variação de repetitividade – CVr, dividido pelo desvio-padrão relativo de repetitividade – DPRrH ou coeficiente de variação de repetitividade – CVrH estimado através da equação de Horwitz, na qual w é a fração de massa do analito (com ambos, numerador e denominador, nas mesmas unidades), e usando a hipótese sr = 2/3 sR:
Incerteza de Medição: Parâmetro não negativo que caracteriza a dispersão dos valores atribuídos a um mensurando, com base nas informações utilizadas [22,28].
Incerteza de Medição Analítica – IMA: Incerteza de medição associada ao mensurando obtido através de um procedimento de análise química validado.
Incerteza de Medição Expandida ou Incerteza Expandida: Produto de uma incerteza padrão combinada com um fator maior do que o número um.
International Union of Pure and Applied Chemistry – IUPAC: União de Química Pura e Aplicada.
Limite de Decisão para Confirmação – CCα: É o limite a partir do qual se pode concluir que uma amostra é não conforme com uma probabilidade de erro α. Sendo que o valor 1 – α significa certeza estatística em porcentagem de que o limite permitido foi excedido.
Limite de Detecção – LD: A menor concentração ou massa do analito que pode ser detectada (mas não quantificada) em uma amostra.
Limite de Quantificação- LQ: A menor concentração do analito que pode ser quantificada. É comumente definido como a concentração mínima do analito na amostra que pode ser determinada com precisão e veracidade aceitáveis sob as condições declaradas do método.
Limite Máximo de Resíduo – LMR: é a concentração máxima de um dado resíduo admissível em determinada matriz.
Limite Máximo de Contaminante – LMC: é a concentração máxima de um dado contaminante admissível em determinada matriz.
Limite de Referência - LR: é o teor máximo admissível de uma determinada substância em uma determinada matriz; estabelecido por legislação nacional ou internacional, pelo cliente interno ou externo.
Linearidade: Capacidade de um método de análise, dentro de uma determinada faixa, de fornecer uma resposta ou resultados instrumentais, proporcionais à quantidade de analito a ser determinada na amostra de laboratório.
Lote: Ver “Batelada”.
Material de Referência Certificado – MRC: Material de referência, acompanhado por um certificado, com um ou mais valores de propriedades, e certificados por um procedimento que estabelece sua rastreabilidade à obtenção exata da unidade na qual os valores da propriedade são expressos, e cada valor certificado é acompanhado por uma incerteza para um nível de confiança estabelecido.
Matriz Representativa: Matriz escolhida para representar um grupo de matrizes de composição semelhante e usada durante a validação de um procedimento analítico. Para qual se tenha demonstrado que não há diferença significativa do resultado do analito quando medido na matriz representativa e na matriz representada.
Método Analítico: Sequência lógica de operações, descritas genericamente e resumidamente, usadas na execução de uma análise química de um dado analito ou um conjunto de analitos em uma dada matriz usando uma dada técnica analítica.
Método Multirresíduo: Método analítico capaz de identificar e quantificar vários tipos de analitos de um mesmo grupo químico ou de grupos químicos distintos, em uma determinada matriz.
Método Qualitativo: Também conhecido como método de “Triagem”: Um método que atende a critérios predeterminados para detectar a presença, ou ausência, de um analito ou classe de analitos, na concentração mínima de interesse ou acima dela.
Método Quantitativo: Método capaz de produzir resultados de concentração do analito com veracidade e precisão que atendam aos critérios estabelecidos.
Não Permitido para a Cultura - NPC: Agrotóxicos de uso registrado no Brasil, porém com o uso não autorizado para a cultura vegetal em questão.
Nível de Interesse ou Nível Requerido: A concentração da substância a analisar numa amostra que é significativa para determinar a sua conformidade com a legislação.
Padrão Interno (surrogate): Composto puro ou elemento adicionado à amostra de ensaio, cujo comportamento químico-físico é considerado como representativo do analito nativo. Às vezes, como nas técnicas analíticas envolvendo espectrometria de massa, são usadas moléculas do analito marcadas com algum isótopo de seus elementos constitutivos.
Padrão de Referência: Padrão, geralmente tendo a mais alta qualidade metrológica disponível em um dado local ou em uma dada organização, a partir do qual as medições executadas são derivadas.
Plano Nacional de Controle de Resíduos e Contaminantes – PNCRC.
Portabilidade: é a possibilidade de o procedimento analítico ser transportado/transferido para outro local de execução ou outro equipamento sem, no entanto, perder suas características metrológicas e seu desempenho.
Precisão de Medição (Precisão): Grau de concordância entre indicações ou valores medidos, obtidos por medições repetidas, no mesmo objeto ou em objetos similares, sob condições especificadas.
A precisão de medição é geralmente expressa numericamente por indicadores de incerteza tais como: dispersão, desvio-padrão (s), variância (s2), desvio padrão relativo (DPR), ou coeficiente de variação (CV), sob condições de medição especificadas. Uma menor precisão é indicada através de um elevado desvio-padrão.
Referência para Tomada de Medidas - RTM: é a concentração de referência para tomada de mediadas legais, aplicável às substâncias proibidas ou não autorizadas.
Recuperação (aparente): conhecida como recuperação aparente, é a proporção de analito determinada no ponto final do método. Geralmente expresso como uma porcentagem em relação à quantidade do analito esperada na amostra.
Valor relacionado à capacidade de um método analítico medir um mensurando corretamente, quando uma quantidade conhecida de mensurando é adicionada à amostra.
Recuperação absoluta: Recuperação real de um analito após etapas de extração e purificação. A proporção de analito (rendimento) restante no ponto da determinação final do método, quando comparado com a concentração esperada da amostra.
Recuperação da concentração retrocalculada (Rcr (%)): estima relação percentual do valor de x calculado/predito (xc) de um ponto de uma curva de calibração, com relação ao valor de x esperado (xi), sendo xc a concentração obtida pelo sinal (yi).
Recuperação da concentração retrocalculada média por nível de calibração (RN (%)): estima o valor médio de Rcr (%) de um nível de calibração, com relação ao valor de x esperado.
Regra de Decisão: regra que descreve como a incerteza de medição é considerada ao declarar a conformidade com um requisito especificado.
Repetitividade de Medição ou Repetitividade (r): Precisão de medição sob um conjunto de condições de repetitividade.
As condições de repetitividade são aquelas em que todos os fatores que podem alterar o resultado da medição são mantidos constantes ou controlados. Isto é, mesmo procedimento de medição, mesmo analista, mesma temperatura, pressão e umidade ambientes, mesmo laboratório, mesmos fornecedores e lotes de reagentes, e replicatas de medição realizadas em curto período.
Reprodutibilidade de Medição ou Reprodutibilidade (R): Precisão de medição sob um conjunto de condições de reprodutibilidade.
As condições de reprodutibilidade são aquelas em que todos os fatores que podem alterar o resultado da medição são variados na maior extensão possível que se pode ter na rotina. As replicatas de medição são feitas em dias diferentes em intervalo de tempo abrangente, em laboratórios, instrumentos e analistas diferentes, entre outros.
Reprodutibilidade interna: Estimativa da reprodutibilidade de um método em um mesmo laboratório, também conhecida como reprodutibilidade intralaboratorial.
Responsável Técnico: Responsável pela unidade laboratorial, o qual é o responsável pela assinatura do Relatório de Ensaio.
Resposta Instrumental: É a indicação (valor indicado, leitura, sinal) do instrumento de medição quando uma amostra de ensaio ou uma solução de calibração é inserida no instrumento de medição, e.g.: absorbância, transmitância e área de pico cromatográfico.
Resultado de Medição: Conjunto de valores atribuídos a um mensurando, completado por todas as outras informações pertinentes disponíveis.
Robustez: Susceptibilidade de um procedimento analítico a alterações das condições experimentais, as quais podem ser expressas como uma lista dos materiais da amostra, das substâncias a analisar, das condições de armazenamento, das condições ambientais e/ou de preparação da amostra em que o método pode ser aplicado tal como apresentado ou com pequenas alterações específicas. Relativamente a todas as condições experimentais que possam, na prática, estar sujeitas a variações (por exemplo, estabilidade dos reagentes, composição da amostra, pH, temperatura), devem ser indicadas quaisquer alterações susceptíveis de afetar os resultados analíticos.
Propriedade de um procedimento analítico que indica sua insensibilidade a mudanças de condições operacionais conhecidas sobre o resultado do procedimento e, portanto, sua adequação a seu propósito de uso.
Raiz Quadrada da Média – RQM: do inglês root mean square (RMS), também conhecida como “Valor Justo”, é a raiz quadrada da média aritmética dos quadrados dos valores.
Seletividade: Propriedade de um sistema de medição, utilizado com um procedimento de medição especificado, segundo a qual o sistema fornece valores medidos para um ou vários mensurandos, tal que os valores de cada mensurando sejam independentes uns dos outros ou de outras grandezas associadas ao fenômeno, corpo ou substância em estudo.
É a extensão na qual um procedimento analítico pode determinar analito(s) particular(es) em uma mistura(s) ou matriz(es) sem a interferência de outros componentes de comportamento semelhante.
Tendência: Ver “Viés”.
Teor Mínimo Calibrado - TMC: a concentração mais baixa a que o sistema de medição foi calibrado.
Validação: Verificação na qual os requisitos especificados são adequados para um uso pretendido.
Veracidade ou Veracidade de Medição: É o grau de concordância entre o valor médio obtido de uma grande série de resultados de ensaio e um valor de referência aceito.
Verificação Concorrente: Forma de Validar ou Verificar um método utilizando os dados gerados no procedimento de controle de qualidade na rotina analítica, é a chamada verificação concorrente (on going) da performance do método.
Viés: Tradução do termo em inglês Bias, sendo a diferença entre um resultado de um ensaio e um valor de referência aceito, expressando a tendência do método. Em termos da estimativa da incerteza de medição analítica, ela corresponde ao erro sistemático. O Viés relaciona-se com a veracidade, quanto maior a Tendência (Viés) menor é o grau de concordância da veracidade com resultado da medição.
Violação (resultado): Resultado analítico em que um resíduo ou contaminante tenha excedido o limite legal (ex: LMR, TMC e RTM).
¶ 1.2. Conceitos
Não aplicável
¶ 2. Responsabilidades
Os parâmetros e critérios descritos neste documento devem ser observados por todos os laboratórios pertencentes à Rede Nacional de Laboratórios Agropecuários, credenciados para determinação de resíduos e contaminantes em alimentos.
O laboratório que pretende solicitar credenciamento ou ampliação de escopo, na área de RCA, também deve atender aos requerimentos presentes neste Manual.
Alterações nos procedimentos, parâmetros ou critérios estabelecidos poderão ser necessários devido à especificidade de determinados procedimento analítico ou peculiaridade do laboratório. Nestes casos, modificações poderão ser consideradas desde que sejam adequadamente justificadas, garantindo a sua consistência científica e conceitual com os princípios deste Manual e o não comprometimento da rastreabilidade, da comparabilidade e da confiabilidade dos resultados analíticos gerados.
O presente manual possui vigência e prazo indeterminado e será revisado sempre que necessário pela Coordenação Geral de Laboratórios Agropecuários do Departamento de Serviços Técnicos (CGAL/DTEC).
A gestão desse manual está sob a responsabilidade da CGAL/DTEC que prestará auxílio ao público alvo leitor. Dúvidas e/ou sugestões quanto à aplicação deste manual devem ser submetidas ao Departamento responsável.
A publicação e atualização das versões na plataforma oficial da SDA para acesso pelo público alvo será de responsabilidade da Secretaria representada pelo DTEC.
¶ 3. Objetivo e Introdução
¶ 3.1 Objetivo
Este Manual foi elaborado com o objetivo de estabelecer parâmetros e critérios de aceitação aplicáveis à validação de métodos analíticos e à rotina analítica dos laboratórios públicos e privados que pertencem ou desejam pertencer à Rede Nacional de Laboratórios Agropecuários, para realização de análises para área de Resíduos e Contaminantes em Alimentos (RCA). A validação dos procedimentos analíticos, segundo os requerimentos deste Manual, visa garantir a validade dos resultados, conferindo-lhes rastreabilidade, comparabilidade e confiabilidade.
¶ 3.2 Introdução
Quando a primeira edição do Manual de Garantia da Qualidade Analítica foi publicada em 2011 foi um enorme avanço para toda rede de laboratórios envolvidos na área de análise de resíduos e contaminantes em alimentos no Brasil, havia uma necessidade eminente de uma padronização de conceitos e procedimentos técnicos bem como o alinhamento com referências nacionais e internacionais.
A publicação do manual conseguiu alinhar de forma bastante aprofundada, todo arcabouço técnico e estatístico relacionados à análise de resíduos e contaminantes em alimentos, tanto no que diz respeito à garantia da validade dos resultados, quanto ao controle de qualidade na aplicação dos métodos na rotina analítica.
Um trabalho árduo já havia se iniciado para o Brasil, em especial para o MAPA, para traçar estratégias de modo a atender os diversos mercados consumidores internacionais, que sempre buscam equivalência dos sistemas de controle sanitário e a segurança dos alimentos e insumos agropecuários. Equivalência que obrigatoriamente passa pelos Laboratórios Oficiais do MAPA que têm o dever de demonstrar que o controle dos resíduos e contaminantes em alimentos está equivalente ao dos mercados consumidores.
Ao longo de mais de uma década de aplicação dos conceitos do manual na rotina analítica, de diversos laboratórios oficiais, inúmeros documentos de referência para a área de RCA foram atualizados e publicados, demonstrando a necessidade de uma nova edição.
Além disso, nesta segunda edição boa parte das atualizações foram realizadas a aproveitando de toda experiência prática adquirida pela equipe técnica do MAPA, em especial dos LFDAs, que utilizaram este manual para guiar os trabalhos analíticos que ocorreram desde a primeira edição, aplicando os conceitos e avaliando como o fluxo analítico tem se comportado. Há uma necessidade de aproveitar essa experiência prática e tornar o manual mais claro e objetivo, condizente com a realidade de um laboratório de rotina analítica em Resíduos e Contaminantes em Alimentos (RCA), mas ao mesmo tempo mantendo a excelência técnica já estabelecida.
Do ponto de vista tecnológico muito se avançou, sistemas instrumentais se tornaram mais sensíveis, seletivos e robustos, como consequência o número de analitos analisados teve um aumento quase exponencial, aproveitando-se de toda tecnologia recém disponível e atendando o crescente número de substâncias analisadas.
Do mesmo modo, a aplicação dos métodos na rotina analítica, sempre acompanhados de ferramentas de controle de qualidade, forneceu dados robustos com relação à acurácia (precisão e veracidade) e à incerteza da medição dos métodos utilizados.
Com a dedicação exemplar de diversos colaboradores, atuantes como responsáveis técnicos nos seis LFDAs espalhados pelo Brasil, e a supervisão da Coordenação Geral de Laboratórios Agropecuários (CGAL) é que esta segunda edição foi elaborada, o empenho de todos foi fundamental para a elaboração e manutenção dessa literatura em estado da arte.
Nélio Fleury Filho
¶ 4. Procedimentos
¶ Parte I - Introdução e Requisitos Gerais
¶ I.1 Evitando Contaminações
O laboratório e as áreas afins (recepção, armazenamento de amostras, armazenamento de reagentes, lavagem de vidrarias e instrumentos, sala de preparo e extração, descarte em geral) necessitam ser planejados tendo em vista diminuir ao máximo a possibilidade de contaminações.
Procedimentos operacionais devem ser escritos de forma a padronizar todas as operações realizadas, contemplando desde a recepção das amostras até a emissão dos resultados.
Reagentes e soluções de altas concentrações devem ser manipulados em locais distintos daqueles onde são preparadas as soluções de baixa concentração. Na impossibilidade deve ser demonstrada, por intermédio de controles, a ausência de contaminação cruzada.
Especial atenção deve ser dispensada ao sistema de exaustão utilizado, de forma a minimizar os riscos de contaminação.
O controle de insetos e roedores, no laboratório e em suas proximidades, somente poderá ser efetuado se devidamente autorizado pelo responsável do laboratório e deve ser feito sob seu controle, de preferência, sem a utilização de substâncias químicas voláteis ou altamente impregnantes. Para laboratório de análise de resíduos de agrotóxicos não é recomendada a realização de Dedetização devido ao alto risco de contaminação cruzada com o princípio ativo utilizado.
¶ I.2 Materiais de Referência Certificados (MRC) utilizados como Padrões Analíticos
Na validação e na rotina analítica devem ser utilizados materiais de referência certificados (MRC) produzidos de acordo com o guia ABNT NBR ISO 17034 [2].
Os MRCs devem ser utilizados sempre que forem comercialmente disponíveis, na ausência de MRC comercialmente disponível, podem ser utilizados Materiais de Referência, desde que estes contenham as informações necessárias conforme recomenda a ABNT ISO GUIA 35 [1].
Os Materiais de referência certificados – MRC, materiais/padrões de referência, devem ser armazenados em local seco, quando necessário protegido da luz, de preferência em baixas temperaturas e sempre sob condições controladas e monitoradas, visando diminuir as taxas de degradação, contaminação ou perdas por evaporação. As condições de armazenamento prescritas pelos fornecedores dos MRC e demais padrões devem ser estritamente observadas.
¶ I.3 Cadeia de Custódia dos Materiais de Referência Certificados (ou Materiais de Referência)
Os MRC devem compor os padrões de trabalho (analíticos) utilizados no preparo da curva de calibração (curva analítica) e no preparo das amostras fortificadas, utilizadas na validação do método e na rotina analítica.
Ao ser adquirido o MRC deve ser avaliado quanto ao atendimento aos critérios da ABNT NBR ISO 17034, bem como a sua adequação técnica para então ser aprovado ao uso.
Os padrões de trabalho (analíticos) devem possuir cadeia de custódia, para permitir total rastreabilidade sobre seu sobre seu recebimento, uso, avaliação e aprovação.
As seguintes informações devem constar da cadeia de custódia:
- Código de identificação (CAS);
- Identificação da substância (IUPAC, DCB/DCI, nome comum);
- Condição de temperatura de armazenamento
- Prazo de validade;
- Pureza;
- Incerteza do material;
- Quantidade adquirida; e
- Avaliação do padrão.
¶ I.4 Preparação, Uso e Armazenamento de Soluções Padrão
O processo de preparação das soluções padrão deve ser devidamente registrado, identificando os componentes e as respectivas quantidades. As soluções devem ser devidamente rotuladas de forma inequívoca e datadas, acondicionadas em recipientes adequados e hermeticamente fechados, para evitar perdas de solventes voláteis e contaminações, e armazenadas à temperatura adequada, se possível abaixo de 10 ºC.
A preparação das soluções padrão a partir de um MRC, deve ser baseada em sua massa, usando balanças com resolução de pelo menos 0,01 mg, deve-se evitar pesar massas de padrões inferiores a 10 mg.
Ao se trabalhar com MRC de sais (cloridrato, sulfato, hidrato, ...) avaliar a necessidade de corrigir a massa levando em conta apenas a base livre da substância, conforme for o caso, ex: Cloridrato de Ractopamina, Massa Molecular do Sal (MMS): 337.8 g/mol, Massa Molecular da Base Livre (MMBL): 301,4, Fator de correção (MMS / MMBL) = 1,121.
A estabilidade de um padrão de referência existente, ao ser repreparado, deve ser verificada comparando as respostas do detector entre a solução antiga e a recém preparada. A comparação deve ser realizada usando diluições apropriadas de padrões individuais ou misturas de padrões. Diferenças inexplicáveis nas concentrações aparentes entre o antigo e o novo padrões devem ser investigados. Discrepâncias entre as concentrações de novas e antigas soluções podem ser devido a uma série de fatores além da simples degradação do analito, e.g. precipitação do analito, evaporação do solvente, diferenças nas purezas entre o antigo e o novos padrões de referência, erros na pesagem, erros no preparo, ou erros na análise instrumental.
O estabelecimento dos prazos de validade deve fazer parte dos procedimentos de validação do método analítico, caso o laboratório opte em trabalhar com armazenamento de soluções.
Admite-se que a determinação do prazo de validade seja conduzida de forma concorrente à rotina analítica caso haja evidências de uma estabilidade adequada em períodos curtos (i.e. 30 a 90 dias)
Maiores detalhas sobre o estudo de estabilidade de soluções padrão são encontrados na Parte III deste manual.
Onde existir evidência suficiente, tais como dados de outros LFDAs, ou referências cientificas e normativas, de que um determinado analito é estável nas condições de armazenamento especificadas (tempo, solvente, temperatura, etc.), então o laboratório que reproduzir essas condições de armazenamento pode utilizar tais informações para determinar a validade da solução padrão preparada. Contudo, a possível ocorrência evaporação do solvente deve ser verificada.
¶ I.5 Reagentes e suas Soluções
As especificações técnicas dos reagentes analíticos devem ser compatíveis com a finalidade de seu uso, de forma a evitar a ocorrência de contaminações e/ou interações que venham interferir na qualidade dos resultados analíticos.
Os reagentes devem ser preferencialmente armazenados em suas embalagens originais, nas condições preconizadas pelo fabricante. O laboratório deve manter controle efetivo sobre o estoque dos reagentes.
¶ I.6 Recepção, Armazenamento e Preparação de Amostras
As amostras recebidas no laboratório devem ser avaliadas em relação aos aspectos de inviolabilidade e adequabilidade do recipiente de contenção, identificação, conservação e quantidade encaminhada. Estando as amostras de acordo com os requisitos de recepção, elas devem ser cadastradas e receber um código, numérico ou alfanumérico, ao qual serão associados, no mínimo, às seguintes informações:
- Origem;
- Data de recepção;
- Estado de conservação;
- Quantidade; e
- Tipo de amostra (especificação).
Após a recepção, as amostras devem ser encaminhadas para a preparação, antes da análise, ou armazenadas de acordo com as suas peculiaridades e requisitos específicos.
Critérios para recebimento de amostras devem seguir prioritariamente os requisitos específicos para o atendimento aos planos oficiais do MAPA.
¶ I.7 Desenvolvimento e Otimização do Procedimento Analítico
O desenvolvimento e a otimização de um procedimento analítico não constituem sua validação em si, mas a precedem. Quando feito pelo laboratório, essas atividades trazem muita expertise na execução do procedimento analítico e muitas informações úteis para o planejamento e condução da validação.
A validação de um procedimento analítico sem sua prévia otimização Intralaboratorial, pode resultar no não atendimento dos requisitos de validação e perda de tempo, trabalho e recursos do laboratório.
A otimização interna do procedimento analítico inclui as calibrações dos instrumentos de medições, a verificação (qualificação) da adequação desses instrumentos e das instalações do laboratório ao procedimento analítico. Assim como a verificação da adequação dos reagentes e demais insumos analíticos e o treinamento dos analistas, por exemplo.
Na otimização do procedimento analítico é feito um estudo dos efeitos dos diversos fatores experimentais que podem afetar no resultado analítico, procurando-se estabelecer as condições experimentais que produzam um resultado com veracidade e precisão adequados ao propósito.
As informações obtidas durante a otimização do procedimento analítico constituem um estudo prévio da robustez do método e que poderá ser utilizado para tal finalidade.
Quando for necessário desenvolver um procedimento analítico para uma combinação analito-matriz (um dado analito em uma dada matriz) de interesse, normalmente inicia-se por uma avaliação de artigos científicos. Algumas vezes é possível encontrar uma metodologia que pode ser aplicada integralmente, mas normalmente é necessário realizar algumas adequações para a realidade do laboratório. Outras vezes, a revisão científica possibilita que o laboratório estabeleça uma metodologia própria de forma célere, reduzindo custos com testes e desenvolvimento. Em todos os casos, a metodologia deve atender aos requisitos de validação deste procedimento antes de ser adotada pelo laboratório.
¶ I.8 Identificação e confirmação do analito por técnicas de cromatografia acoplada à espectrometria de massas
A espectrometria de massa acoplada a um sistema de separação cromatográfica é uma ferramenta muito poderosa para identificação de um analito na amostra. Fornece simultaneamente dados de tempo de retenção, relações massa/carga (m/z) e a abundância relativa (intensidade) dos íons, a combinação dessas informações é capaz de confirmar a presença do analito com altíssimo grau de confiança.
¶ I.8.1 Requisitos para a separação cromatográfica
Para a cromatografia líquida (LC) ou gasosa (GC), recomenda-se que o tempo de retenção mínimo aceitável para o(s) analito(s) em estudo deve ser o dobro do tempo de retenção correspondente ao volume morto da coluna. O tempo de retenção do analito no extrato deve corresponder ao do padrão de calibração, de um padrão em extrato de matriz ou de um padrão de matriz fortificada, com uma tolerância de ± 0,1 minuto. Caso se utilize um padrão interno, a razão entre o tempo de retenção cromatográfica do analito e o do padrão interno, ou seja, o tempo de retenção relativa do analito, deve corresponder ao do padrão de calibração, com um desvio máximo de 0,5% para a cromatografia gasosa e de 1% para a cromatografia líquida.
¶ I.8.2 Requisitos para espectrometria de massas
Devido à alta sensibilidade e seletividade, a espectrometria de massas (MS) se tornou a técnica analítica de escolha na área de resíduos e contaminantes. Assim, uma atenção especial deve ser dada no que se refere aos procedimentos para identificação e confirmação da presença de resíduos e contaminantes nas amostras.
Diversas formas de realizar a identificação e confirmação da presença do analito são descritas em documentos internacionais e em referências bibliográficas, a depender do tipo de técnica ou analisador empregado, mas de maneira geral a confirmação pode ser feita pelo espectro de massas, massa exata, pelo padrão isotópico, ou pelo sinal/área de íons selecionados. [6, 14, 36, 55]
Em geral, e especialmente quando se emprega analisadores únicos (não sequenciais), íons de alta razão massa/carga (m/z) são mais seletivos do que íons de baixa m/z (por exemplo, m/z <100). No entanto, íons resultantes da perda de água ou de fragmentações comuns podem ser pouco seletivos e devem ser, sempre que possível, evitados. Embora os íons isotópicos característicos, especialmente os aglomerados de Cl ou Br, possam ser particularmente úteis, os íons selecionados não devem originar-se exclusivamente da mesma parte da molécula do analito. Íons moleculares, moléculas (des)protonadas ou adutos são altamente característicos do analito e devem ser incluídos no procedimento de medição e identificação, sempre que possível. A escolha dos íons para identificação pode mudar dependendo da interferência de fundo (background). Nesses casos, a identificação de um analito depende da correta seleção dos íons utilizados, eles devem ser suficientemente seletivos para o analito na matriz analisada e na faixa de concentração relevante.
A utilização de íons (ou fragmentos) selecionados tem-se tornado a forma mais comum e objetiva para a identificação/confirmação da presença de um analito na amostra, em especial quando se utiliza analisadores do tipo triplo quadrupolos. Nessa abordagem o monitoramento seletivo de reações (SRM, selected reaction monitoring), tem-se mostrado a forma mais utilizada para área de resíduos e contaminantes, sendo selecionadas duas transições SRM para cada analito, geralmente, a transição mais sensível e/ou seletiva é utilizada para quantificação enquanto uma segunda transição é utilizada para confirmação da presença do analito. A identificação do analito é feita através da intensidade relativa do íon no espectro ou da área do pico cromatográfico correspondente ao íon extraído. Nos cromatogramas, os picos cromatográficos correspondentes aos íons empregados na confirmação do analito devem necessariamente se sobrepor em mesmo tempo de retenção.
A razão dos valores de intensidade ou área correspondente aos íons monitorados do analito (o de menor intensidade dividido pelo de maior intensidade), deve corresponder àquelas dos padrões em extrato de matriz, ou dos padrões em matriz fortificada ou das soluções padrão em solvente, em concentrações comparáveis, medidas nas mesmas condições. Pequenos desvios na razão dos íons são aceitáveis, conforme a recomendação específica de cada área podendo ser aceitos valores de desvio entre 15 e 40%.
A avaliação dos valores de desvio da razão de íons visa demonstrar a seletividade da técnica de análise por espectrometria de massas, já que se observa a capacidade de distinção assertiva da identidade do analito quantificado. Tal característica do método é avaliada na etapa de validação do método, que será abordado com maiores detalhes na Parte II do manual.
Outra forma de promover a identificação e confirmação do analito na amostra por MS é empregando medidas de massa exata. A espectrometria de massas de alta resolução (HRMS) é usualmente empregada para descrever equipamentos que apresentam poder de resolução superior a 10.000 (full width at half maximum - FWHM). Para confirmação da identidade de um analito em HRMS, recomenda-se avaliação de ao menos dois íons diagnósticos com exatidão de massa inferior a 5 ppm. Vale ressaltar a importância da avaliação da especificidade dos íons de escolha, mesmo em HRMS. O alto poder de resolução não apenas aumenta a exatidão de massas, mas também reduz o risco de sobreposição de picos cujas massas exatas sejam muito próximas. Porém, existem casos em que equipamentos com maior poder de resolução podem ser necessários para discriminar íons cujas massas exatas podem ser afetadas devido a presença de interferentes de matriz.
Para maior confiança na identificação, podem ser obtidas mais evidências a partir de informações adicionais de espectrometria de massa. Por exemplo, avaliação de espectros de varredura completa (full scan), padrão de isótopos, íons de adução ou íons produtos (em MS/MS) adicionais.
¶ I.9 Validação de Procedimentos Analíticos
A validação é um estudo experimental e documentado que objetiva demonstrar que o procedimento analítico avaliado é adequado à finalidade proposta, de forma a assegurar a confiabilidade dos resultados obtidos.
É essencial que a validação seja realizada sobre o procedimento analítico exatamente da forma que ele será executado na rotina do laboratório.
O planejamento, a preparação e a execução da validação, devem seguir protocolos de validação detalhados contemplando:
- Adequação ao uso pretendido: finalidade e âmbito de aplicação;
- Responsável técnico do projeto;
- Pessoal técnico envolvido com as respectivas responsabilidades;
- Identificação das Unidades, equipamentos/instrumentos utilizados;
- Documentos, tais como instruções de trabalho e de método, inicial de execução do procedimento analítico para pré-validação;
- Parâmetros de desempenho e critérios de aceitação;
- Experimentos de pré-validação e de validação propriamente dita;
- Características de desempenho dos equipamentos/instrumentos;
- Qualificação dos materiais (padrões, reagentes, amostras, alíquotas, entre outros);
- Realização dos experimentos da pré-validação: dados (registros) e conclusão;
- Realização dos experimentos da validação: Dados (registros) e conclusão;
- Descrição do Método final e definitivo para a execução do procedimento analítico na rotina;
- Relatório Final de Validação.
Em se tratando de método normalizado, os parâmetros críticos (veracidade e precisão) devem ser avaliados com o intuito de demonstrar que os procedimentos preconizados atendem aos critérios de aceitação estabelecidos neste Manual. Caso sejam realizadas alterações no procedimento normalizado, a validação deverá ser conduzida na extensão necessária.
¶ I.10 Conduções do Processo de Validação
A validação de um procedimento analítico constitui o primeiro nível e primeira ferramenta da garantia da validade de um sistema de qualidade laboratorial.
Segundo a ABNT NBR ISO/IEC 17025:2017 [3] a validação de um método é a confirmação por exame e fornecimento de evidência objetiva de que os requisitos específicos para um determinado uso pretendido são atendidos. A própria Coordenação Geral de Acreditação do Inmetro (Cgcre) possui um documento de caráter orientativo DOQ-CGCRE-008 rev. 09, intitulada “orientação sobre validação de métodos analíticos” [39], o qual tem como objetivo auxiliar os laboratórios de ensaio a verificar a adequação do método ao propósito.
Documentos de referência internacionalmente aceitos sobre validação de métodos, tais como o Guia IUPAC/ISO/AOAC [46] e o Guia EURACHEM [49], também foram amplamente utilizados na elaboração deste manual.
Para a área de RCA, de maneira geral não existe a disponibilidade de métodos normalizados, de modo que a maioria dos métodos utilizados devem ter a adequação ao proposito verificada através da sua validação.
Dentre os documentos e guias internacionais que tratam do desenvolvimento e validação de métodos da área RCA podemos destacar aqui os seguintes:
Resíduos de Medicamentos Veterinários:
- Regulamento de Execução (UE) 2021/808 da Comissão de 22 de março de 2021. Relativo ao desempenho dos métodos analíticos para os resíduos de substâncias farmacologicamente ativas utilizadas em animais produtores de géneros alimentícios e à interpretação dos resultados, bem como aos métodos a utilizar na amostragem, e que revoga as Decisões 2002/657/CE e 98/179/CE [33].
Resíduos de Agrotóxicos em Alimentos:
- European Commission Document SANTE 11312/2021 v2. Analytical quality control and method validation procedures for pesticide residues analysis in food and feed [19];
- Codex Alimentarius Guideline CXG/GL 40-1993:2010. Guidelines on good laboratory practice in pesticide residue analysis [9]; e
- Codex Alimentarius Guideline CXG 90-2017. Guidelines on performance criteria for methods of analysis for the determination of pesticide residues in food and feed [13].
Contaminantes Inorgânicos em Alimentos (inclui HPAs):
- Regulamento (CE) 333/2007 da Comissão de 28 de março de 2007 que estabelece métodos de amostragem e de análise para o controlo oficial dos teores de chumbo, cádmio, mercúrio, estanho na forma inorgânica, 3-MCPD e benzo(a)pireno nos géneros alimentícios [30].
Contaminantes Orgânicos em Alimentos (Dioxinas e Furanos, e PCBs):
- Regulamento (UE) 2017/644 da Comissão de 5 de abril de 2017 que estabelece métodos de amostragem e análise para o controlo dos teores de dioxinas, PCB sob a forma de dioxina e PCB não semelhantes a dioxinas em determinados géneros alimentícios e que revoga o Regulamento (UE) nº 589/2014 [31].
Contaminantes Biológicos (Micotoxinas):
- Regulamento de Execução (UE) 2023/2782 da Comissão de 14 de dezembro de 2023 que estabelece os métodos de amostragem e de análise para o controlo dos teores de micotoxinas nos géneros alimentícios e que revoga o Regulamento (CE) n° 401/2006 [34];
- European Commission Document SANTE/12089/2016 - Guidance document on identification of mycotoxins in food and feed [20]; e
- European Commission Document EURL-MP-guidance doc_003 (version 1.4) Guidance document on performance criteria for methods of analysis for mycotoxins and plant toxins in food and feed (draft 17 May 2024) [27].
Neste tópico iremos abordar os parâmetros fundamentais que são comuns ao procedimento de validação de métodos de RCA, as instruções aqui descritas devem ser utilizadas sempre em complemento aos critérios específicos exigidos de cada área de atuação (Parte VI do manual), são eles descritos abaixo:
- Faixa de Trabalho;
- Linearidade; e
- Efeito Matriz.
- Veracidade (Recuperação/Tendência);
- Precisão (Repetibilidade e Reprodutibilidade Interna);
- Limites de Interesse:
- Limite de Detecção;
- Limite de Quantificação; e
- Limite de Decisão (CCα) e Capacidade de Detecção (CCβ).
- Seletividade;
- Razão de Íons; e
- Robustez.
Para minimizar a carga de trabalho do processo de validação, recomenda-se que seja adotado um planejamento cuidadoso e estatisticamente consistente, que permita combinar ensaios de forma a determinar diferentes parâmetros de validação em um mesmo experimento.
Os experimentos de validação podem ser otimizados com o objetivo de se obter respostas que serão utilizadas para estimar diversos critérios em paralelo. Por exemplo, o estudo de linearidade pode ser combinado com o efeito matriz, do mesmo modo podem ser combinados os experimentos para avaliação da veracidade e precisão, sendo que a partir deles é possível se obter os diferentes tipos de limites de interesse, bem como a seletividade do método e a avaliação da razão de íons.
¶ Parte II - Parâmetros e Critérios de Aceitação de Desempenho de um Procedimento Analítico
¶ II.1 Níveis de Interesse Analítico
Os parâmetros e os critérios de validação aplicáveis às determinações dos analitos (resíduos ou contaminantes) devem levar em consideração o nível de concentração de interesse, exemplos mais comuns são: Limite Máximo de Resíduo (LMR), Limite Máximo de Contaminante (LMC), nível de referência para tomada de medida (RTM), Limite de Quantificação (LQ), Teor Mínimo Calibrado (TMC), ou outro nível de concentração no qual alguma decisão será tomada.
¶ II.2 Linearidade e Faixa de Trabalho
¶ II.2.1 Considerações gerais
Linearidade (ou regressão linear) é a capacidade de o método produzir resultados diretamente proporcionais à concentração do analito na amostra, dentro de um intervalo especificado. De uma forma mais ampla, a linearidade é a capacidade de o procedimento analítico produzir curvas de calibração que podem ser adequadamente ajustadas pela equação de uma função linear nos parâmetros e que são capazes de realizar predições com confiabilidade.
A técnica instrumental de análise direciona inicialmente a forma de se construir uma curva de calibração, de modo que a resposta instrumental se comporte de forma linear frente variações da concentração do analito (ou da quantidade do analito).
Dentre as técnicas de análise mais utilizadas na área de RCA destaca-se a espectrometria de massas, em especial a CLAE-EM/EM, seguida da CG-EM/EM e por último as técnicas utilizadas na análise de elementos inorgânicos tais como EAA e ICP-MS. Todas eles têm em comum o comportamento linear entre a resposta instrumental e a quantidade de analitos.
Na maioria dos casos instrumentais descritos acima a faixa linear se comporta como regressão linear de primeiro grau,
Y = a + b . x, onde
Y = Resposta instrumental;
x = Concentração;
a = componente do intercepto da regressão.
b = componente da inclinação (ângulo) da regressão.
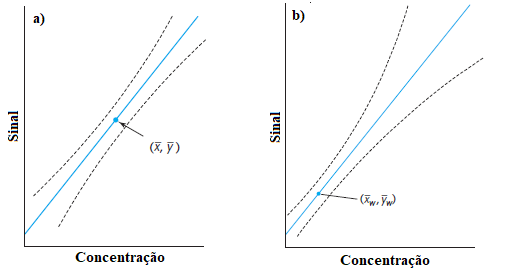
Desta forma a regressão pode ser facilmente estimada pelo Métodos dos Mínimos Quadrados Ordinários (MMQO), este que encontra a curva que minimiza os valores dos resíduos de cada ponto da regressão conforme a técnica de cálculo de minimização [58].
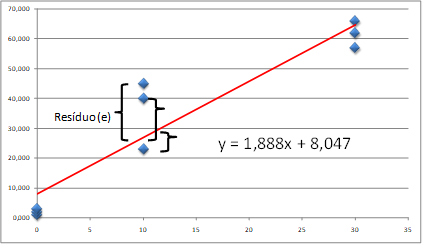
Na Figura 1 observa-se graficamente os resíduos de três pontos de uma curva de calibração, quanto maior o valor absoluto do resíduo de um ponto, pior é o seu ajuste à curva de calibração.
O valor do resíduo (e) de cada ponto é estimado conforme a fórmula abaixo:
Resíduo (e) = yi – Yci,
Onde:
Yci = (a + b . xi);
Yci = y calculado para o xi;
xi = concentração correspondente ao ponto yi;
yi = sinal instrumental obtido pelo ponto xi;
O modelo MMQO é bastante conhecido e estudado, dada a sua simplicidade e eficiência para aplicação em diversas situações, entretanto, matematicamente o MMQO somente pode ser aplicado quando os dados se comportam de forma homocedástica, ou seja, o desvio padrão de Y se mantém constante ao longo da faixa de valores de x. Esta condição nem sempre está presente nos dados experimentais, dependendo do alcance da faixa linear e da técnica de análise utilizada.
O que se observa na maioria dos casos é que desvio padrão de Y aumenta na medida que o valor de Y se eleva, ou seja, os dados se comportam de maneira heterocedástica. O comportamento heterocedástico fica mais evidente em faixas lineares com alta magnitude de alcance.
Para avaliar a homoscedasticidade dos resíduos de uma curva de calibração podem ser realizados testes de significância, tais como o teste de Brown-Forsythe (ou teste de Levene modificado), ou o teste F (Fischer-Snedecor), dentre outros. [59].
A heteroscedasticidade também pode ser visualmente observada ao traçar o gráfico do desvio padrão de cada nível de calibração em função da concentração, caso o valor de desvio padrão de um nível se mostre muito diferente de um outro nível (i.e., diferenças maiores que três vezes) indicam que os dados não são homocedásticos.
Essa falta de ajuste ao MMQO ocorre, pois, o método trata todos os dados de maneira igual (ordinária), a curva de calibração obtida de fato é o resultado dos menores valores dos resíduos, ou seja, os pontos de maior valor de Y tem maior peso no momento de definir a regressão.
Ao mesmo tempo, dependendo da sensibilidade do analito frente o detector, a faixa de concentração se torna muito ampla e os pontos de maior concentração fogem da faixa linear de 1o Grau, como esses pontos têm uma influência muito grande no MMQO eles acabam por prejudicar consideravelmente o ajuste e a incerteza da curva de calibração.
Em análises químicas dados heteroscedásticos normalmente são tratados utilizando regressões ponderadas, de modo que os pontos medidos com maior precisão (menor variâncias), influenciam mais os parâmetros da adequação da função do que os com precisão inferior. Para esse propósito a quantidade que minimiza os parâmetros de adequação dos mínimos quadrados é ajustada através de uma ponderação inversamente proporcional à variância de cada ponto (Si2) conforme for o peso (wi) [52]:
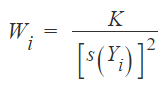
Em que K é uma constante de normalização normalmente escolhida de modo que:
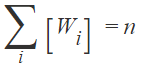
Onde n corresponde ao número total de pontos a serem ajustados.
Outra forma utilizada para estimar Wi, equivalente à descrita acima, é segundo a fórmula abaixo [53]:
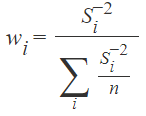
O Método dos Mínimos Quadrados Ponderados – MMQP, como é conhecido, pode ser realizado com diferentes formas de ponderação além das citadas acima, atualmente muitos analistas têm assumido que os dados são hetroscedásticos de modo que Wi varia conforme o inverso de xi, e.g., 1/x e 1/x2 sem a necessidade de estimar experimentalmente variância de Yi para realizar a ponderação.
No Anexo II estão presentes as fórmulas e os cálculos da estimativa dos parâmetros da regressão pelo método dos mínimos quadrados ponderados MMQP elaborado considerando as principais referências bibliográficas [51, 52, 53 e 61], além dos cálculos, o Anexo II apresenta um estudo de caso que compara os diferentes tipos de Regressão Linear (MMQO e MMQP (Σsi2/Isi2 , 1/x e 1/x2)) para a técnica de determinação de micotoxinas em alimentos por cromatografia líquida acoplada à espectrometria de massas dupla (CL-EM/EM).
Como pode ser observado na Figura 2 e no Anexo II, o tipo de método utilizado (MMQO ou MMQP) não tem um grande impacto nos valores de intercepto e inclinação, mas sim na incerteza da curva de calibração, afetando diretamente os limites de confiança do MMQO, que se tornam muito amplos nos níveis de calibração menores, algo que não ocorre para o MMQP. Esse impacto na incerteza é ainda maior para curvas de calibração com amplitude muito grande entre o primeiro e o último nível de calibração [56].
Ressalta-se que atualmente a maioria dos equipamentos já possuem em sua programação a possibilidade de ajuste da regressão por diferentes tipos de ponderação, sendo que o próprio analista pode definir o tipo de ponderação que se adequa melhor aos dados, entretanto, ressalta-se que mesmo que os dados tratados pelo MMQP aparente estar visualmente melhor, a adequação ao modelo deve ser verificada, assim como o impacto do modelo nos limites de confiança da regressão [6].
Como pode ser observado nos estudos de caso dos Anexos II e III, mesmo que o modelo MMQO tenha obtido bons resultados nos testes de adequação ao modelo, ele não deve ser utilizado se os dados têm uma distribuição heteroscedástica.
Uma forma bem objetiva de verificar o impacto da adequação do modelo é avaliar a dispersão dos resíduos dos pontos da curva de calibração, em especial nos níveis de concentração críticos. Essa dispersão é objetivamente observada ao calcular a Recuperação do valor de xic (x calculado/predito) em relação ao xi (esperado ou verdadeiro).
A Recuperação da concentração retrocalculada (Rcr %) de um ponto estima relação percentual do valor de x calculado/predito (xic), com relação ao valor de x esperado (xi), sendo xic a concentração obtida pelo sinal (yi). A forma de estimar a Recuperação da concentração retrocalculada dos pontos da curva de calibração é descrita com maiores detalhes no Item II.2.2, sendo que quanto mais distante de 100% for o valor da Recuperação do ponto maior é o seu erro.
Valores ruins de Rcr (%) indicam que a curva de calibração não está tendo uma boa acurácia, e indica que a quantificação das amostras também será prejudicada.
No Anexo III contém um outro estudo de caso sobre o impacto do uso do MMQO e do MMQP para um mesmo conjunto de dados, produzidos durante a validação de um método para determinação de resíduos de agrotóxicos em alimentos por CLAE-EM/EM, o estudo provê um destaque maior no impacto do modelo nos valores de Rcr (%) dos pontos da curva de calibração e na estimativa da incerteza da regressão.
Como o MMQO é um caso particular do MMQP a utilização deste último é sempre correta e recomendada, eliminando, nesse caso, a necessidade de se fazer o teste de homoscedasticidade da curva de calibração.
Experiência prática com uso de curvas de calibração na área de RCA em cromatografia acoplada à espectrometria de massas indicam que regressões lineares com um alcance muito amplo (i. e. alcance maior que 10x da diferença de concentração entre o primeiro nível e o último nível de calibração) devem ser ajustadas pelo MMQP para que os níveis mais baixos obtenham bons valores de Veracidade.
Testes de normalidade (Ryan-Joiner) e independência (Durbin-Watson) dos resíduos podem ser realizados para auxiliar na avaliação dos parâmetros da linearidade [59].
Para a avaliação da adequação ao modelo linear MMQO, o teste de análise de variância (ANOVA) pode ser utilizada para a avaliação da significância da regressão [51] e falta de ajuste (Item A3 do Anexo II).
Além do teste de ANOVA mencionado, e a avaliação da Recuperação dos pontos da curva de calibração, é recomendado verificar o ajuste através da análise do gráfico dos resíduos, gerado conforme o modelo linear adotado, no qual observa-se o valor do resíduo de cada ponto por nível de concentração, como demonstrado na Figura 3 abaixo, A avaliação visual do gráfico pode fornecer indícios sobre possíveis erros no ajuste linear e a dispersão dos resíduos.
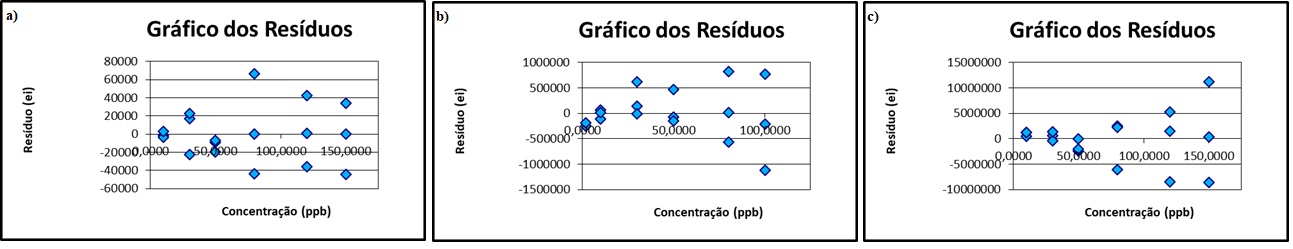
¶ II.2.2 Procedimento prático
A estimativa da linearidade deve ser realizada pelo menos para a curva de calibração que se pretende utilizar durante a validação do método e após isso na rotina analítica.
Essencialmente existem três tipos de curva de calibração que normalmente são utilizadas nos procedimentos analíticos, são elas:
- Curva de Calibração em Solvente (CCS): Curva de Calibração na qual os seus pontos de calibração são preparados a partir de adição de uma solução padrão em um solvente (ou reagente) usual, normalmente esse solvente deve ser o mesmo presente no final das etapas de extração/digestão e purificação do método.
- Curva de Calibração Matrizada (CCM): Curva de Calibração na qual os seus pontos de calibração são preparados a partir de adição de uma solução padrão em um extrato/digerido de uma amostra branca, após as etapas de extração/digestão e purificação. O preparo de tal curva visa avaliar possíveis efeitos matriz e o seu uso em rotina é recomendado para minimizar esses efeitos, entretanto tal preparo de CCM não corrige erros oriundos da recuperação do analito.
- Curva de Calibração em Matriz Fortificada (CCF): Também conhecida como Curva de Calibração de Procedimento, é a Curva de Calibração na qual os seus pontos de calibração são preparados a partir de adição de uma solução padrão em uma amostra branca antes das etapas de extração/digestão. Os pontos são então analisados instrumentalmente após todas as etapas de extração/digestão e purificação. Este tipo de curva de calibração é o que mais simula as condições reais que amostra sofre durante o processo analítico, por esse motivo o uso de tal curva é indicado para minimizar possíveis efeitos matriz e compensar por erros originários da recuperação do analito.
Caso o analista opte por utilizar uma Curva de Calibração em Solvente durante o procedimento de validação e após isso na rotina analítica, ele deve demonstrar a ausência do efeito de matriz, ou que este efeito matriz não seja significativo. A avaliação do Efeito Matriz será tratada mais adiante neste manual.
Caso já se possua conhecimento prévio de que exista efeito matriz, ele pode optar por não realizar o estudo de efeito matriz e utilizar apenas as curvas de calibrações matrizadas, CCM ou CCF, conforme for o caso.
Sendo assim, a linearidade deve ser demonstrada para a curva selecionada.
De uma forma geral, o planejamento de elaboração da curva analítica deve contemplar os seguintes critérios:
- Determinar a faixa de concentração de interesse, a qual deve preferencialmente contemplar o limite de referência utilizado (LMR, LMC, LTM, LQ...).
- Preparar a curva de calibração indicada, com padrão, em no mínimo 5 níveis de concentração (incluindo o ponto 0, caso exista), preferencialmente igualmente espaçadas ou espaçadas de forma a evitar pontos de alavanca, cada nível de curva de calibração deve ser injetado em triplicata, podendo ser triplicata de preparo ou triplicata de injeção conforme for a indicação técnica.
- O ponto zero pode ser utilizado como ferramenta de controle de qualidade, para avaliação do ruído e/ou do zero do instrumento, podendo este representar o nível zero de concentração;
- Recomenda-se que a sequência de injeção (ou análise) dos pontos da curva de calibração ocorra na ordem em que elas são injetadas na rotina analítica, caso não haja uma recomendação o analista deve optar por injetar os pontos em ordem crescente de concentração, sendo injetado ponto de cada nível de concentração por vez, conforme exemplo de sequência na Figura 4 abaixo. Caso seja necessário avaliar a independência dos resíduos da curva de calibração, recomenda-se que a injeção dos pontos pode ocorra de forma aleatória [59].
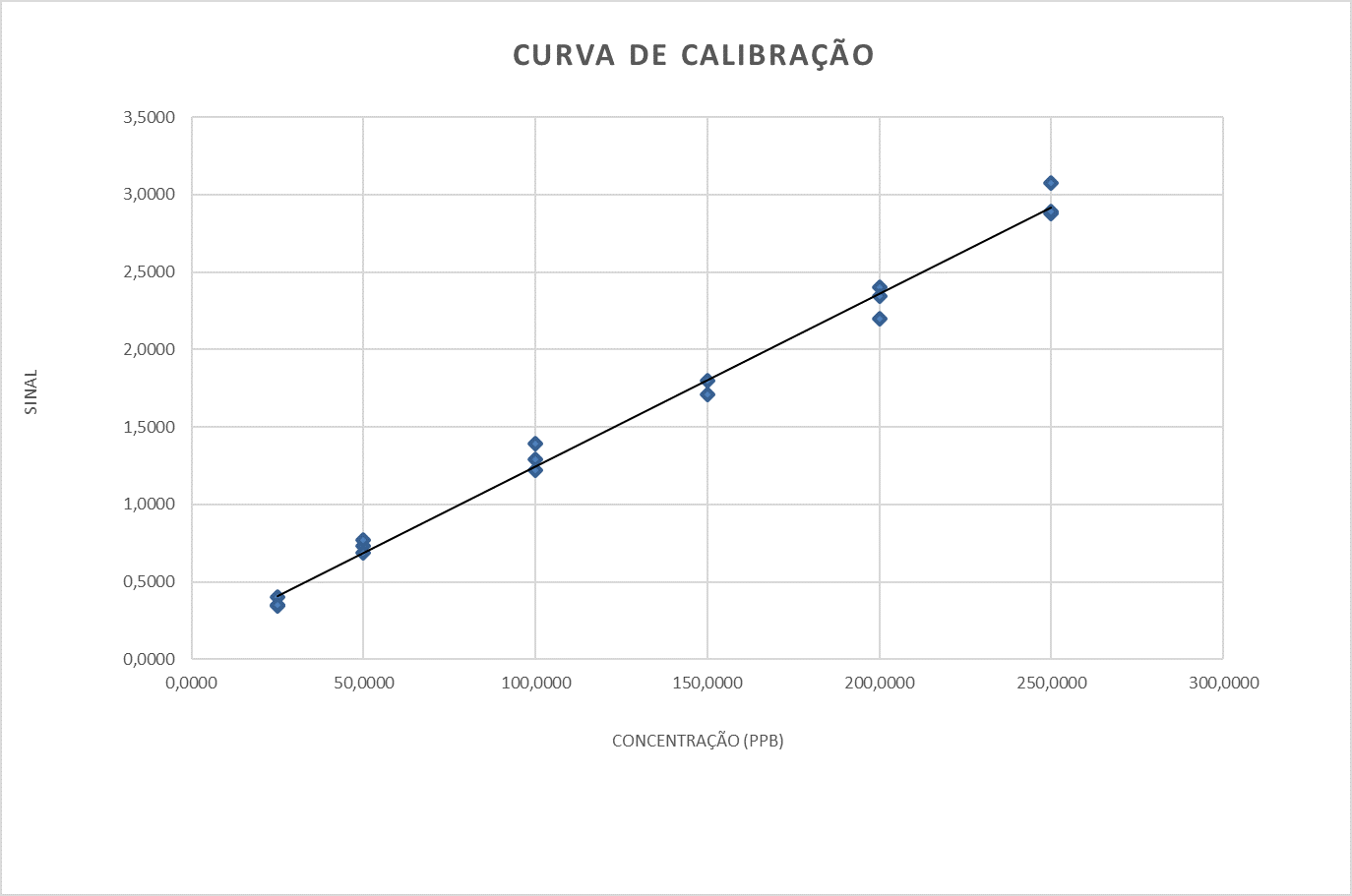
Verificar na Figura 5 abaixo um exemplo de como deve ser plotada a curva de calibração, atentando ao fato de que todos os pontos utilizados devem estar presentes:
Estimar então os seguintes valores que serão utilizados para verificar a adequação ao modelo proposto:
- Coeficiente de Correlação (r);
- Recuperação da concentração retrocalculada (Rcr %) de cada ponto da curva de calibração, estima relação percentual do valor de x calculado/predito (xc) com relação ao valor de x esperado (xi), sendo xc a concentração obtida pelo sinal (yi).
Rcr (%) = xic * 100 / xi
Sendo:
xic = concentração calculada na curva de calibração a partir de yi, sendo xic = (yi – a)/b;
xi = Valor esperado para x, ou valor verdadeiro de x;
- Recuperação média da concentração retrocalculada por Nível (RN %), estimado como sendo a Rcr (%) média dos pontos por nível de calibração, conforme descreve abaixo:
RN j = MÉDIA (Rj,1; Rj,2; Rj,n; ...), onde
Rj,n = Recuperação (Rcr %) no ponto n no nível j.
No Anexo II e III estão presentes exemplos de valores obtidos para os parâmetros citados para o juste linear por MMQO e MMQP.
Testes para valores anômalos (outliers) podem ser aplicados nos pontos da curva de calibração testado, dentre esses testes destacamos os três abaixo, entretanto outros testes podem ser aplicados, recomenda-se que dos valores anômalos identificados sejam excluídos no máximo 22,4% dos pontos, sem excluir um nível inteiro:
- Teste de Grubbs ou Teste Q de Dixon [5], para os valores de Rcr (%) dos pontos da curva de calibração;
- Teste do resíduo padronizado Jacknife [59];
¶ II.2.3 Critérios de Aceitabilidade
Os critérios de aceitabilidade para os parâmetros da curva de calibração serão definidos na Parte VI do manual, conforme for área específica de RCA (Parte VI do manual).
¶ II.3 Efeito Matriz
A diferença relativa entre a resposta instrumental de um analito em solvente e ele em matriz é chamada de Efeito Matriz (EM).
A avaliação do efeito de matriz demanda uma comparação entre as respostas instrumentais obtidas do analito na presença e na ausência dos componentes da matriz presente no extrato (ou digerido) final das amostras.
Objetivamente podemos estimar o valor relativo do Efeito Matriz conforme a seguinte fórmula:
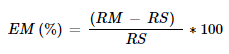
Em que:
RM = Resposta instrumental do analito puro em extrato de matriz branca,
RS = Resposta instrumental do analito puro em solvente.
Valores de EM positivos indicam que a presença da matriz resultou em ganho de resposta instrumental quando comparado com o analito puro em solvente, do mesmo modo, valores de EM negativos indicam a ocorrência de supressão da resposta instrumental.
A fim de se obter a resposta instrumental do analito puro em extrato de matriz branca a fortificação do analito na matriz branca deve ocorrer após a etapa de extração / digestão e purificação da amostra, visando desconsiderar o impacto da recuperação do analito no estudo de Efeito Matriz.
O efeito matriz deve ser então avaliado apenas se o método faz (ou pretende fazer) o uso de Curva de Calibração em Solvente (CCS), para que seja demonstrado que a quantificação das amostras não está sendo prejudicada pela ocorrência do Efeito Matriz.
Em espectrometria de massas, em especial na Cromatografia Líquida a ocorrência de efeito de matriz é bastante conhecida, ele envolve o fenômeno de ganho ou supressão de resposta instrumental devido à presença de componentes da matriz, que ocorre na fonte do Espectrômetro de massas [6].
Já para Cromatografia Gasosa acoplada à espectrometria de massas o EM ocorre principalmente pela interação dos componentes da matriz com a fase estacionária da coluna cromatográfica, o que afeta a retenção dos analitos e o seu perfil cromatográfico, e por consequência, influencia a resposta instrumental [19].
Para técnicas óticas (Espectrometria de Absorção Atômica, ICP-OES, ...) a resposta instrumental pode ser prejudicada devido à presença de componentes da matriz que afetam a capacidade de atomização dos metais durante a etapa de emissão ou absorção e na etapa de nebulização.
A ocorrência de efeito matriz é bastante comum nos métodos da área de RCA, de modo que a maioria dos métodos utilizam da CCM ou da CCF como uma alternativa para anular, ou minimizar o Efeito Matriz.
A avaliação do EM pode ser realizada de duas formas mais comuns:
1. Comparando duas curvas de calibração: CCS x CCM, neste caso aproveita-se as curvas de calibração avaliadas na etapa da linearidade (conforme Item II.2.2), conforme exemplificado na Figura 6;
- Comparar os valores de intercepto e inclinação, e/ou;
- Comparar os valores médios por nível de calibração;
2. Comparação média em dois níveis de concentração (ex: LQ, LMR, 10x LQ) preparados em solvente e na presença de matriz, podendo ser utilizados níveis correspondentes ao da curva de calibração, neste caso recomenda-se no mínimo triplicata de preparo por nível de concentração comparado.
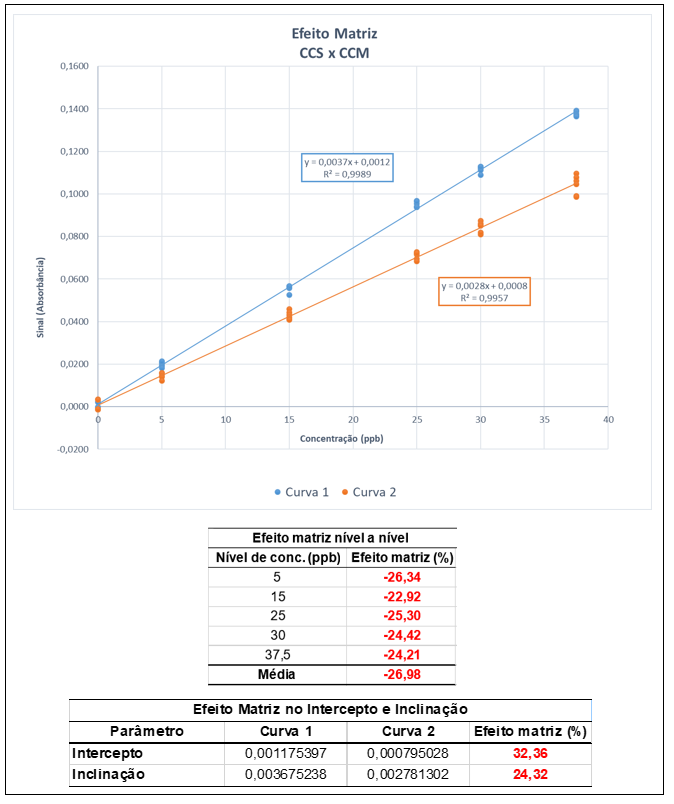
Calcular então o Efeito Matriz, caso o valor seja impactante é indicada a presença de efeito matriz e a possível necessidade de utilização da curva de calibração matrizada (CCM).
Limites máximos para o valor de efeito matriz devem ser verificados conforme recomendação específica para cada área da RCA.
Caso o estudo aponte a presença de efeito matriz o analista pode optar por confirmar a significância da diferença observada através de testes de significância (e.g., teste t para médias) [5].
Caso não haja recomendação específica quanto à um limite máximo para o valor de efeito matriz a sua ocorrência pode ser avaliada pela comparação de médias (nível à nível) através de testes de significância como o teste t para médias.
¶ II.4 Veracidade e precisão do método
¶ II.4.1 Considerações gerais
A avaliação da Veracidade e da Precisão de um método é de fundamental importância para a garantia da validade dos resultados gerados, sendo que a Veracidade está relacionada com os erros sistemáticos e a precisão está relacionada com a componente dos erros aleatórios, estão, portanto, ligados diretamente à incerteza da mediação analítica.
Por esses motivos a avaliação da Precisão e Veracidade de um método deve ser abrangente o suficiente para garantir que o laboratório dispõe de um método validado, adequadamente controlado e com os seus erros devidamente conhecidos.
Os Valores de Veracidade e Precisão do método devem ser demonstrados em condições de reprodutibilidade interna, simulando ao máximo as condições observadas na rotina do laboratório.
Os Experimentos para determinar a Precisão e Veracidade do método podem ser combinados para serem estimados utilizando-se dos mesmos dados experimentais.
A maioria dos experimentos são realizados com a utilização de amostras brancas fortificadas com Solução Padrão do analito em concentrações estabelecidas.
A etapa de fortificação das amostras deve ser realizada no início do procedimento de extração. É necessário que, após a fortificação das amostras brancas, elas sejam homogeneizadas e deixadas em repouso por no mínimo 15 min, de modo a ocorrer a correta ambientação dos analitos com a matriz, sendo que somente após isso é que a extração deve ser iniciada. Tal recomendação visa garantir uma maior representatividade da amostra fortificada frente uma amostra naturalmente contaminada.
¶ II.4.2 Veracidade (Recuperação/Tendência)
O vocabulário internacional de metrologia define a veracidade como: “Grau de concordância entre a média de um número infinito de valores medidos repetidos e um valor de referência” [40], sendo que a Recuperação é a capacidade de um método analítico medir um mensurando corretamente, quando uma quantidade conhecida de mensurando é adicionada à amostra. Trata–se de um meio efetivo para avaliar a Veracidade do sistema analítico, já que testa o método na presença de outros compostos que estejam contidos na mesma matriz da amostra.
Na validação de um método a Veracidade pode ser demonstrada através dos valores de Recuperação (aparente) estimados como a relação percentual médio entre o valor medido e o valor esperado para uma amostra de concentração conhecida, conforme a fórmula abaixo, também conhecida como recuperação aparente.
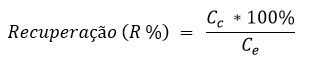
Onde:
Cc = Concentração calculada no analito na amostra;
Ce = Concentração esperada do analito na amostra;
Já o Viés (V), do inglês Bias, indica a Tendência da veracidade do método, representa o desvio relativo em porcentagem da recuperação do método com relação ao valor esperado, o parâmetro é útil na estimativa da incerteza da medição do método e é associado ao seu erro sistemático.
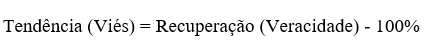
A determinação da Veracidade usualmente é realizada com o uso de amostra branca fortificada, na qual adiciona-se quantidade conhecida do analito (MRC) antes do início do procedimento analítico.
Os valores de Veracidade devem ser demonstrados em diferentes níveis de concentração de interesse, contidos dentro da faixa de trabalho do método, tais como o Limite de Quantificação, LMR, LTA, 10x LQ, e outros, devendo observar as recomendações referentes aos níveis de concentração estudados conforme a Parte VI do manual.
A Veracidade é estimada como a média das recuperações individuais por nível de calibração. A quantidade de dados utilizados para calcular a média vai depender da quantidade de dados utilizados para a estimativa da repetibilidade e/ou reprodutibilidade interna.
Testes para valores anômalos (outliers) podem ser aplicados, tais como os Testes de Grubbs e o Teste Q de Dixon para identificar e excluir valores anômalos.
Assim, deve ser estabelecido o valor de Veracidade médio por nível de concentração. Estes valores devem estar dentro das faixas de aceitabilidade recomendadas para a área específica, conforme definido na Parte VI deste manual.
Caso os valores de Veracidade médios obtidos se situem fora da faixa de aceitabilidade recomenda-se que os resultados sejam corrigidos por essa Veracidade, entretanto essa possibilidade de correção fica condicionada ao fato do analito ter obtido resultados satisfatórios de precisão.
O uso da curva de calibração de matriz fortificada (CCF) é uma boa alternativa para lidar com analitos de recuperação fora da faixa aceitável, já que utilizando esse tipo de curva de calibração os efeitos da recuperação são corrigidos automaticamente pela regressão, sem a necessidade se utilizar de correções matemáticas.
¶ II.4.3 Precisão
É a estimativa da dispersão de resultados entre ensaios independentes, repetidos de uma mesma amostra, amostras semelhantes ou padrões, em condições definidas.
A precisão é estimada sob duas condições de análise: repetibilidade e reprodutibilidade.
Quando a reprodutibilidade envolve um único laboratório ela é chamada de reprodutibilidade intralaboratorial (ou reprodutibilidade interna, ou reprodutibilidade parcial).
A precisão de repetibilidade é demonstrada pela realização de réplicas de análise em uma mesma condição, em um mesmo dia, por um mesmo analista e com os mesmos equipamentos;
Já a reprodutibilidade interna é demonstrada pela realização de réplicas de análise em condições distintas, tais como dias distintos, analistas diferentes, equipamentos diferentes;
¶ II.4.4 Procedimento de determinação da precisão de repetibilidade e reprodutibilidade interna
Preparar e analisar um conjunto de amostras constituídas de amostras brancas fortificadas em dois ou mais níveis de concentração. Analisar réplicas de amostra para cada nível de concentração, o número de réplicas por nível / lote está definido na parte VI deste manual conforme for a área de atuação.
Realizar a análise utilizando as mesmas condições, no mesmo dia, por um analista e utilizando os mesmos equipamentos, insumos e regentes.
Para a reprodutibilidade interna realizar o procedimento descrito acima em diferentes ocasiões, alterando as condições da batelada de análise, tais como diferentes dias, instrumentos de medição e/ou analistas.
Calcular a concentração encontrada para cada amostra fortificada.
Estimar a precisão de repetibilidade e reprodutibilidade interna para cada nível de concentração, com base nos valores de desvio padrão de repetibilidade (sr) e o desvio padrão de reprodutibilidade interna (sR). Esses valores podem ser calculados conforme descrito na Tabela 1, em linha com o que é recomendado pelo documento ISO 5725-3 [42] e equivalente ao procedimento recomendado no Anexo C do documento EURACHEM [49] o qual utiliza-se da ferramenta ANOVA.
Tabela 1: estimadores das variâncias de repetibilidade (sr2) e reprodutibilidade interna (sR2) para uma dada concentração.
.png)
Tal média ponderada pode ser calculada da seguinte forma:
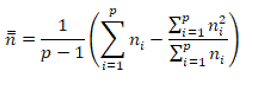
Em que:
p corresponde ao número de bateladas (e.g., p = 3).
Assim, obtém-se a variância entre bateladas Sb2, que posteriormente pode ser combinada à variância repetibilidade (Sr2) para obter a variância de reprodutibilidade (interna) SR2, conforme for atendida as condicionais:
- Se Sb2 for menor que Sr2 o valor de SR será o mesmo do Sr2,
- Se Sb2 for maior ou igual que Sr o valor de SR considera a combinação de Sb2 e Sr2;
O valor de Sr2 descrito acima corresponde à chamada variância combinada, na qual é realizada uma combinação ponderada dos valores de variância de cada dia da batelada [53].
O valor de SR também pode ser calculado da forma direta, na qual se calcula o desvio padrão de todos os dados por nível de fortificação, neste caso um cuidado deve ser tomado quando o valor de Sr for maior que o valor de SR, caso isso ocorra, recomenda-se que o SR2 adote o valor do Sr, em consonância com o que é realizado no procedimento descrito na Tabela 1.
Após o cálculo de Sr e SR, podem ser calculados os desvios padrão relativos de repetibilidade (DPRr) e de reprodutibilidade interna (DPRR), respectivamente, dividindo cada desvio padrão pela média dos resultados de um dado nível.
Na Tabela 2 abaixo está um exemplo de cálculos de precisão e veracidade conforme o documento ISO 5725-3.
Tabela 2: Exemplo de resultados de precisão e veracidade (recuperação) estimados em um nível de concentração de 10 µg/kg. Resultados de concentrações de desvios padrão estão todos em µg/kg.
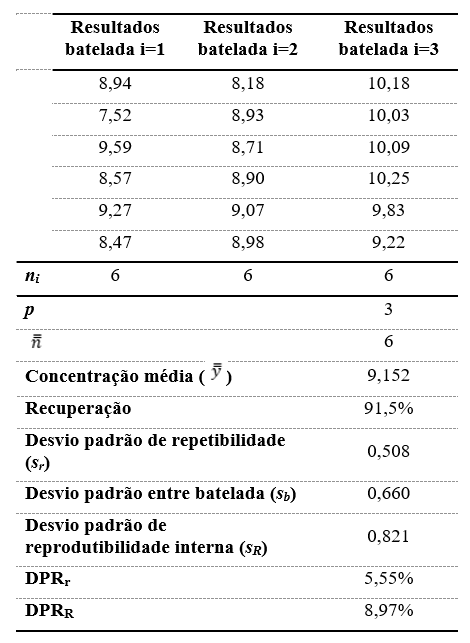
Testes para valores anômalos (outliers) podem ser aplicados, tais como os Testes de Grubbs e o Teste Q de Dixon para identificar e excluir valores anômalos.
¶ II.4.5 Critérios de Aceitabilidade para Veracidade e Precisão
A aceitabilidade dos valores de Veracidade e precisão deve ser verificada conforme for indicado na área de atuação presente na Parte VI deste manual.
¶ II.5 Limites de Interesse
A área de RCA tem em comum análises que devem ser capazes de obter resultados com Veracidade e Precisão adequados nos níveis de contração de interesse para tomada de ação regulatória, concentrações que normalmente se situam em níveis traço, na ordem de partes por bilhão.
Portanto, o estabelecimento dos limites de Interesse de um método é uma forma de garantir a validade dos resultados situados entorno desses limites.
Dentre os termos mais comuns destacamos aqui o Limite de Detecção (LD), Limite de Quantificação (LQ), Limite de Decisão (CCα) e Capacidade de Detecção (CCβ), sendo que esses dois últimos tem uma aplicação específica para área de resíduos de medicamentos veterinários e serão explicados com maiores detalhes na Parte VI.
¶ II.5.1 Limite de Detecção – LD
Em termos gerais, o limite de detecção (LD) é a menor quantidade ou concentração de analito na amostra de teste que pode ser distinguida de zero com segurança. [46]
Esse termo é definido no VIM 2012 [40] como: "Valor medido, obtido por um dado procedimento de medição, para o qual a probabilidade de declarar falsamente a ausência de um componente em um material é β (Taxa de falso negativo), sendo α a probabilidade de declarar falsamente a sua presença (taxa de falso positivo). ”
Apesar da aparente simplicidade do conceito, ao aprofundar no assunto uma série de situações devem ser consideradas, o que restringe o uso de tal limite em análises oficiais de resíduos e contaminantes em alimentos. Desta forma, conceito de LD tem sido utilizado quase exclusivamente para métodos qualitativos (ou métodos de triagem). Logo, o estabelecimento do LD para métodos quantitativos não é obrigatório.
Diversas abordagem poder ser utilizadas para estabelecer matematicamente o valor de LD, por exemplo:
- A concentração que corresponde a três vezes a relação sinal x ruído;
- A concentração correspondente a três vezes o desvio padrão do resultado da análise de amostras brancas (ou amostras com baixo teor do analito);
- A concentração correspondente três vezes o desvio padrão do menor nível da curva de calibração.
Para métodos qualitativos o valor do LD necessita ser validado experimentalmente, correspondendo à concentração em que a taxa de falso negativo seja menor ou igual a 5%. O estabelecimento do Limite de Detecção para métodos qualitativos está descrito no Item II.9.
¶ II.5.2 Limite de Quantificação – LQ
Várias definições e diferentes procedimentos são apresentados na literatura e em diferentes normas internacionais.
Muitas das abordagens citam formas matemáticas de se estabelecer o LQ, tais como a concentração correspondente a 10 vezes a relação sinal x ruído, ou a concentração correspondente à 10 vezes o desvio padrão do resultado da análise de amostra branca (ou amostra com baixo teor do analito), ou a concentração correspondente ao menor nível da curva de calibração.
Independente de como foi estimado o valor de LQ, este deve sempre ser verificado na prática durante o procedimento de validação do método, de modo que seja demonstrado que no LQ o método possui precisão e veracidade adequados.
Assim, como uma forma de otimizar os experimentos de validação de métodos, o LQ do método pode ser estabelecido como o menor nível de concentração validado no estudo de Precisão e Veracidade.
¶ II.5.3 Limite de Decisão – CCα e Capacidade de Detecção – CCβ
São parâmetros definidos na Decisão 808 [33] que medem o desempenho do procedimento analítico, levando em consideração a incerteza da medição, no nível de concentração no qual se toma alguma decisão, o chamado nível de interesse. Para maiores detalhes com relação ao estabelecimento desses limites verificar a o Item VI.1.
¶ II.6 Seletividade
Seletividade é o grau em que um método pode identificar o analito na presença de interferentes. Idealmente, a seletividade deve ser avaliada para qualquer interferente importante que possa estar presente.
Diversos experimentos podem ser utilizados para demonstrar a seletividade do método, a maioria envolve a análise de amostras brancas de diferentes origens e avaliar na resposta instrumental a ocorrência de interferentes (pico cromatográfico, sinal, traço de íon...) na região onde se espera a resposta do analito.
Quando apropriado, é recomendável fortificar amostras brancas com substâncias que possam interferir na identificação e/ou quantificação da substância de interesse, em concentrações relevantes. Em seguida, deve-se avaliar a ocorrência de interferência no resultado.
Sugere-se uma seletividade mínima de 30% de interferência com relação Limite de Quantificação do método.
Para métodos qualitativos a seletividade do método está relacionada à taxa de falso positivo, devendo esta ser inferior à 5% para que o método seja considerado adequado. Maiores detalhes com relação à verificação da seletividade para métodos qualitativos estão descritos no Item II.9.
¶ II.7 Razão de Íons
Tratando de métodos de detecção por cromatografia acoplada à espectrometria de massas, a seletividade do método pode ser demonstrada pelos critérios de desempenho aplicados à técnica, conforme discutido no Item I.9.
Como demonstrado, a técnica de cromatografia/espectrometria de massa demanda um número definido de pontos de confirmação para identificar a presença de um analito na amostra. Esses pontos só são atingidos quando os íons utilizados na detecção são observados. No caso do modo SRM (Single Reaction Monitoring), duas transições SRM são necessárias: uma para a quantificação do analito e outra para a qualificação (identificação).
A relação entre os sinais das duas transições é denominada razão de íons. Essa razão de íons é uma característica identificadora do analito. Portanto, ao comparar o valor da razão de íons na amostra com o valor nos padrões, é possível confirmar a identidade do analito na amostra com elevado grau de certeza.
Na rotina analítica a confirmação da presença do analito nas amostras através da avaliação razão de íons é obrigatória para os casos em que o analito foi quantificado acima do limite legal, sendo que a diferença nos valores de razão de íons não deve ser superior à um valor estabelecido conforme a área de atuação, para área de resíduos e agrotóxicos admite-se um desvio máximo de 30%, enquanto que para medicamentos veterinários o valor pode ser de até 40%, os valores específicos para cada área estão descritos na Parte VI.
Portanto, a avaliação da adequação da razão de íons está diretamente relacionada à seletividade do método validado. Já que essa avaliação representa mais uma ferramenta empregada para confirmar a presença do analito, permitindo distinguir possíveis interferentes e evitar falsos positivos.
Logo, a seletividade da razão de íons deve ser avaliada durante a validação do método, como parte do estudo de seletividade, para isso deve ser realizada a comparação das razões de íons obtidas nas amostras fortificadas com relação às da curva de calibração, preferencialmente por nível de concentração, podendo ser utilizados os dados do estudo de Precisão e Veracidade. O valor da média da diferença, deve ser inferior ao limite para variação da razão de íons.
Uma forma alternativa para avaliar a razão de íons é estimar o valor do DPR (%) das razões de íons das amostras fortificadas utilizadas no estudo de Precisão e Veracidade, e / ou com os pontos utilizados na curva de calibração no estudo da linearidade. O valor de DPR (%) deve ser inferior ao limite para variação da razão de íons.
¶ II.8 Robustez
O estudo da robustez de um procedimento analítico procura avaliar o quão sensível o resultado analítico é frente as variações nas condições experimentais do procedimento analítico, os chamados fatores ou tratamentos ou grandezas de influência [40].
Demonstra-se então a estabilidade do método sob diferentes condições, tais como; diferentes condições ambientais, diferentes fabricantes de insumos, colunas cromatográficas, pequenas variações na proporção dos reagentes utilizados, temperatura, agitação, dentre outros.
A avaliação da robustez pode ser realizada na etapa de otimização do método, ou mesmo na etapa de pré-validação, fazendo o uso de ferramentas de planejamento experimental e técnicas de planejamento fatorial [5], um dos testes fatorais mais utilizados em química analítica é o chamado teste de Youden que constitui um método confiável para avaliação da robustez de métodos analíticos, tal planejamento é recomendados por diversas referências em RCA [30 33] para avaliar a robustez de um método. Este teste ocorre por meio de um delineamento que envolve sete parâmetros analíticos combinados em oito experimentos, no Anexo IV está presente um exemplo da aplicação do teste de Youden para a determinação de contaminantes inorgânicos em alimentos.
Outra forma de se avaliar a robustez de um método é pela realização do acompanhamento dos resultados das amostras de controle de qualidade durante a rotina analítica, de modo que seja demonstrado que os valores de Precisão e Veracidade estão se mantendo adequados ao longo do tempo. Tal forma utiliza-se dos dados de controle de qualidade interna e ferramentas de carta controle.
Esta última forma de avaliar a robustez é indicada para métodos de análise de resíduos em alimentos [19 e 33], já que fazendo uso do planejamento fatorial, a quantidade excessiva de respostas obtidas para cada analito dificulta a interpretação dos resultados, pois a resposta pode variar de acordo com a característica do analito frente os diferentes fatores utilizados.
¶ II.9 Métodos Qualitativos
Métodos Qualitativos, também chamados de métodos de Triagem (screening) tem sido cada vez mais utilizado por laboratórios com o intuito de elevar a capacidade operacional do laboratório e otimizar a aplicação de métodos quantitativos, já que as amostras são inicialmente analisadas de forma qualitativa e, sendo identificada a presença do analito na amostra, ela é então analisada por um método quantitativo.
A técnica é especialmente útil quando a maioria das amostras apresenta resultado não quantificado, ou não detectado, assim não sendo necessário analisa-las quantitativamente.
Quanto aos parâmetros a serem avaliados em métodos qualitativos, se convencionou que dois parâmetros devem ser obrigatoriamente avaliados:
Seletividade: verificar Item II.6, normalmente é estimado pela análise de 20 ou mais amostras brancas, as quais devem gerar uma taxa de falso positivo máxima de 5% das amostras analisadas (ou seja, no máximo 1 falso positivos de um total de 20 amostras);
Limite de Detecção, ou Capacidade de Detecção (CCβ): Verificar Item II.5.1, normalmente é estimado pela análise de 20 ou mais amostras brancas fortificadas na concentração de interesse (LD), as quais devem gerar uma taxa de falso negativo máxima de 5% das amostras analisadas (ou seja, no máximo 1 falso negativo de um total de 20 amostras).
Para a área de Resíduos de Medicamentos Veterinários o termo Capacidade de Detecção (CCβ) é utilizado como equivalente ao LD, conforme rege o documento CIR (EU) 2021/808 e o guia “EURL Guidance Document on Screening Method Validation” [22 e 33].
Maiores detalhes para método qualitativos para a área de resíduos de medicamentos veterinários vão estar presentes na parte VI no manual.
¶ II.10 Expressão e Interpretação dos Resultados de Medições
Os resultados das concentrações de cada analito em cada amostra devem ser acompanhados de suas incertezas e da unidade de medição conforme o formato: “Nome ou símbolo do mensurando = valor da concentração do analito ± incerteza expandida (normalmente estimada no nível de confiança de 95%, k = 2) [62].
Ao utilizar um resultado para decidir se ele indica conformidade ou não frente uma especificação, é necessário levar em conta a incerteza de medição. Para isso deve ser definido pelo laboratório a regra de decisão que descreve como a incerteza da medição é considerada ao declarar a conformidade frente um requisito especificado [66]. A ISO/IEC 17025 também exige que, quando relevante, a regra de decisão a ser utilizada seja acordada com o cliente [38].
A regra de decisão do laboratório deve conter claramente procedimentos que envolvem pontos críticos relacionados com o resultado emitido, tais como [62]:
- O número necessário de medições replicadas (se houver) e o procedimento para usar resultados replicados, incluindo (por exemplo) se os resultados devem estar de acordo individualmente ou se deve ser calculada a média antes da comparação com os limites;
- Procedimentos para lidar com valores anômalos (outliers);
- Procedimentos para ações futuras, para uma regra de decisão não binária, quando a decisão é condicional (aprovado/reprovado);
- O procedimento a ser seguido no caso de um resultado condicional que exija medições adicionais;
Ao ser aplicada uma regra de decisão quanto à conformidade de uma amostra, considerando a incerteza do método, duas abordagens são utilizadas na área de RCA conforme for a atuação:
- O resultado somente é considerado não conforme quando o valor quantificado na amostra diminuído da incerteza expandida do método se encontra acima do Limite Legal (e.g., LMR, LQ para agrotóxicos NPC e agrotóxicos proibidos), conforme está exemplificado na Figura 8a, tal abordagem é utilizada na área de resíduos de agrotóxicos em alimentos:
- O resultado somente é considerado não conforme quando o valor quantificado se encontra acima do LMR somado da incerteza expandida do método no LMR, ou quando o resultado quantificado se encontra acima do CCα para a área de Resíduos de Medicamentos Veterinários (RMV), conforme exemplificado na Figura 8b, abordagem bastante comum para contaminantes (orgânicos e inorgânicos) e micotoxinas em alimentos e, equivalente ao adotado em RMV.
A definição quanto à aplicação a regra de decisão depende da indicação do demandante.
¶ II.11 Relatório de Validação
Após a validação do método, um documento deve ser redigido no qual os desempenhos alcançados pelo procedimento validado são relatados. Esse documento deve conter o maior número possível de informações, fornecendo evidências para demonstrar a qualidade do procedimento validado e sua adequação ao uso pretendido.
Neste relatório dissertativo devem estar claramente descritos os parâmetros de validação estudados e os resultados obtidos, bem como uma avaliação sobre o cumprimento dos critérios de aceitação para cada um dos parâmetros estudados.
O relatório de validação não se resume apenas aos dados brutos, razão pela qual se recomenda a utilização do modelo de relatório de validação estabelecido pela CGAL presente no Anexo V no qual contém a estrutura padrão do relatório, bem como as informações mínimas que devem estar presentes;
Os dados brutos (cromatogramas, curvas de calibração, sequência de injeção, dados da análise, ...) devem estar prontamente disponíveis, caso esses dados estejam disponíveis apenas eletronicamente, tal fato deve estar descrito, o local de armazenamento deve ser registrado estando ele controlado e seguro.
¶ II.12 Incerteza da medição analítica (IMA)
¶ II.12.1 Aspectos gerais
O Vocabulário internacional de metrologia (VIM) [40] define a incerteza como um “parâmetro não negativo que caracteriza a dispersão dos valores atribuídos a um mensurando, com base nas informações utilizadas”
O conhecimento da IMA é essencial para a interpretação dos resultados. Sem a avaliação quantitativa da IMA é impossível decidir se a diferença observada entre dois resultados representa mais do que uma variação experimental, se existe adequação de uma especificação, ou até mesmo se limites legais têm sido descumpridos. Sem a informação da incerteza há o risco de ocorrer a má interpretação do resultado. A tomada de decisão incorreta quanto à interpretação do resultado pode gerar despesas desnecessárias na indústria, nos procedimentos legais e consequências adversas em questões sociais.
Em se tratando especialmente dos métodos de análise utilizados na área de Resíduos e Contaminantes em Alimentos (RCA), a aplicação da IMA é uma necessidade. As análises na maioria das vezes visam verificar a conformidade de um produto perante um padrão regulatório, portanto a aplicação da incerteza pode influir diretamente na tomada de decisão quanto à conformidade da amostra.
A norma ABNT ISO/IEC 17025:2017 define com relação à estimativa da IMA: os laboratórios de ensaio devem identificar as contribuições significativas e devem estimar a incerteza da medição para todos os métodos de ensaio.
Diversas formas de se estimar a IMA têm sido propostas nas últimas duas décadas. A documentação fundamental para a estimativa da incerteza é o ISO Guide to Expression of Uncertainty in Measurement, GUM JCGM 100 e 101:2001 [46 e 47]. Entretanto, na Europa o Guia EURACHEM/CITAC Quantifying Uncertainty in Analytical Measurement [16] tem sido bastante utilizado na química analítica por ter uma linguagem mais acessível.
Além desses dois documentos, diversos outros têm sido publicados, como o procedimento NORDTEST [50] e o procedimento ISO 21748:2017 [45], dentre outros [7 e 28], com diferentes modelos para estimativa da IMA, entretanto até o atual momento não existe um protocolo único de estimativa universalmente aplicado [35].
Uma discussão completa sobre a incerteza (de medição) está além do escopo deste Manual.
Ressalta-se aqui dois documentos publicados pelo CODEX Alimetarius alinhados com a aplicação para a área de Resíduos e Contaminantes em Alimentos. Primeiramente o documento CXG 54-2004:2021 Guidelines on Measurement Uncertainty [10], que cobre aspectos gerais da incerteza da medição em análise quantitativa, com definições e terminologias associadas e descreve a importância na interpretação do resultado para avaliação da adequação em planos de amostragem e inspeção de lotes.
Um segundo documento é o CXG 59-2006:2011 Guidelines on Estimation of Uncertainty of Results [11]. Este documento foi elaborado levando em consideração as recomendações gerais do Codex Committee on Methods of Analysis and Sampling (CCMAS), tem uma abordagem ligada diretamente à área de Resíduos, em especial para a de resíduos de agrotóxicos em alimentos.
Apesar do documento CXG 59-2006:2011 ter o foco na área de resíduos de agrotóxicos em alimentos, do ponto de vista técnico os mesmos conceitos podem ser aplicados para as diversas áreas de resíduos e contaminantes em alimentos, pois as técnicas de análise são semelhantes e os níveis de concentração são próximos, assim espera-se que a estimativa da IMA possa ser equivalente.
A escolha da abordagem apropriada depende do tipo de medição ou análise, do método utilizado, do nível de confiabilidade exigido e da urgência da solicitação de uma estimativa da incerteza de medição. Em geral, os procedimentos baseiam-se em abordagens conhecidas como “bottom-up” ou “top-down”.
A abordagem top-down (TD) é baseada em dados de validação de métodos e em dados de precisão de longo prazo derivados de análises laboratoriais, amostras de controle de qualidade, resultados de testes de proficiência, dados de literatura publicada e/ou colaboração interlaboratorial.
A abordagem bottom-up (BU), é feita componente por componente e incorpora um processo baseado em atividades em que o analista divide todas as operações analíticas em atividades primárias. Estes são então combinados ou agrupados em atividades comuns e uma estimativa feita da contribuição dessas atividades para o conjunto valor de incerteza do processo de medição. A abordagem BU pode ser muito trabalhosa e requer um conhecimento detalhado de todo o processo analítico. O benefício para o analista é que esta abordagem fornece uma compreensão clara das atividades analíticas que contribuem significativamente para a incerteza de medição e que, portanto, podem ser atribuídas como pontos de controle críticos para reduzir ou gerenciar a incerteza de medição em aplicações futuras do método.
A principal distinção entre as duas abordagens é que enquanto a abordagem BU parte de uma consideração físico-química do mecanismo de medição, a abordagem TD começa a partir de um conjunto de dados em que a variação entre os diferentes resultados de medição é diretamente observável. Nesse sentido, pode-se dizer que a abordagem BU é teórica, enquanto a abordagem TD é empírica [7]. Apesar das diferenças entre as duas abordagens para estimativa da IMA ambas são reconhecidas como ferramentas igualmente válidas e adequadas [16].
As fontes de incerteza podem ser divididas em dois tipos, a Tipo A que envolve o uso de dados de repetição de experimentos, como dados de validação, carta controle, interlaboratoriais, incerteza herdada da curva de calibração, e Tipo B, que correspondem às incertezas que não são do Tipo A, Incertezas herdadas da calibração dos equipamentos (balanças, pipetas, micropipetas, balões volumétricos...), incertezas herdadas dos padrões de referência certificados, efeitos das condições ambientais, dentre outros [61].
Independente da forma como o laboratório vai estabelecer a IMA o primeiro passo é a identificação das principais fontes de incerteza, isto demanda que o analista tenha um conhecimento mais aprofundado da técnica de análise aplicada de modo a identificar as etapas críticas da análise e verificar possíveis meios de controlar, minimizar, ou mesmo eliminar o efeito.
Assim, a estimativa da IMA produz muito mais informação do que apenas o valor da incerteza expandida do método, sendo que a informação obtida no estudo pode ser utilizada para melhorar o desempenho do método gerando resultados mais precisos.
Os componentes típicos de incerteza incluem efeitos associados ao equipamento (gravimétrico e volumétrico), instrumental, analista, matriz da amostra, método, calibração, tempo e condições ambientais, nos quais estão sujeitos a erros sistemáticos e aleatórios.
Identificadas as possíveis fontes de incerteza o laboratório deve optar por adotar pelo menos uma das abordagens recomendadas para o estabelecimento da IMA.
- Modelagem
- Bottom-up: avaliação de componente por componente de acordo com JCGM 100 [47] ou de acordo com JCGM 101 (Método Monte-Carlo) [48].
- Validação Intralaboratorial
- Top-down: de acordo com ISO GUM JCGM 100 [47], Nordtest TR 537 [50] e o Guia EURACHEM/CITAC: Quantifying Uncertainty in Analytical Measurement (incerteza dos resultados obtidos usando o mesmo procedimento em um único laboratório sob condições variadas) [16].
- Validação Interlaboratorial
- Top-down: utilizando o desvio padrão de reprodutibilidade (ISO 5725-2, ISO 5725-3 e ISO 21748) (incerteza dos resultados obtidos utilizando o mesmo procedimento em laboratórios diferentes) [41, 43, 44 e 45].
- Ensaio de proficiência
- Top-down: usando o desvio padrão para avaliação de proficiência (incerteza dos resultados obtidos pela análise da(s) mesma(s) amostra(s) em laboratórios diferentes) [59]
Embora um estudo detalhado deste tipo possa exigir um esforço considerável, é essencial que custo despendido não seja desproporcional. Na prática, um estudo preliminar identifica rapidamente as fontes de incerteza mais significativas e, como mostram os exemplos, o valor obtido para a incerteza combinada é quase inteiramente controlado pelas contribuições principais. Assim, uma boa estimativa da incerteza pode ser feita concentrando esforços nas maiores contribuições [16].
Vale ressaltar que no procedimento bottom-up pode ocorrer de alguma fonte não ser considerada, ou ainda a contribuição das fontes identificadas for mal estabelecida (matematicamente ou teoricamente), gerando um risco maior da IMA não ser estimada na extensão adequada.
O uso do procedimento TD é incentivado, pois em toda rede de laboratórios que atuam em RCA são utilizadas metodologias validadas, que estimam a precisão e veracidade do método, assim as informações necessárias para o cálculo da incerteza TD já estão disponíveis e são originários de dados já aprovados pelos critérios de validação ao qual eles foram testados.
Caso o laboratório opte por estimar a incerteza pelo procedimento bottom-up, ele deve avaliar criticamente do valor encontrado e compará-lo com o que é estimado pelo método top-down, qualquer divergência significativa deve ser investigada.
Mesmo possuindo diversos conceitos envolvidos, assim como as diferentes abordagens matemáticas aplicadas no estabelecimento da IMA, uma rede de laboratórios oficiais deve buscar uma padronização suficiente para tratar os resultados analíticos de forma imparcial, isonômica e estatisticamente comparável.
Aos laboratórios que compõem a Rede Oficial de Laboratórios Agropecuários na área de RCA é recomendado que seja reportada a incerteza calculada pelo procedimento top-down. Caso a incerteza reportada tenha sido estimada pelo método bottom-up, o valor deve ser equivalente ao valor encontrado pelo top-down.
¶ II.12.2 Estimando a IMA pelo modelo Top-Down
Visando a padronização de procedimentos do MAPA define-se a abordagem top-down para o estabelecimento do IMA como recomendação geral para área de RCA, sendo que recomendações específicas para cada área estão presentes na Parte VI deste manual.
O procedimento recomendado descrito abaixo envolve a estimativa da incerteza considerando os erros aleatórios com erros sistemáticos, estes que podem ser correlacionados respectivamente com os dados de precisão e veracidade do método.
A precisão de um método sob condições de reprodutibilidade é estimada pelo valor de Desvio Padrão Relativo de reprodutibilidade (DPRR(%)), enquanto a veracidade do método é expressa em termos da sua Tendência (ou do Viés). O valor de Viés é então calculado como Recuperação (ou veracidade) (%) diminuído de 100%, conforme foram definidos ao longo da Parte II do manual.
Erros sistemáticos devem ser, sempre que possível, minimizados, e quando esses erros são significativos eles devem ser considerados [46], sendo necessária uma análise crítica quanto à possibilidade de ser aplicada uma correção matemática dos resultados pelo Viés.
Caso seja necessário aplicar uma correção do resultado pelo valor do Viés, o laboratório deve ter uma forma de estabelecer esse valor, levando em conta características do método, da matriz, e da sua reprodutibilidade, da faixa de trabalho, dentre outros fatores. Ressalta-se que um valor de Viés mal estabelecido pode adicionar um erro maior do que o ganho em si pela correção.
Se o valor de viés for significativo e não houver a possibilidade de correção matemática dos resultados pela recuperação, o resultado reportado deve considerar a incerteza estimada e o viés do método [16]. Uma das possibilidades de considerar o viés ao reportar um resultado é incluir ele no cálculo da incerteza padrão combinada. Esta possibilidade de incluir o Viés no cálculo da incerteza parte do princípio de tratar todos os componentes de incerteza de forma isonômica, assim evitando ao máximo subestimar a incerteza de um método, porém por outro lado, essa abordagem pode gerar um valor de incerteza muito grande [65], pois passa a tratar o viés como um desvio padrão [63].
O valor da incerteza combinada deve ser, então, calculado de acordo com que o laboratório trata a Recuperação (ou o Viés) do método, duas formas são propostas para calcular a incerteza combinada, uma para os casos em que o método não tem o resultado corrigido pela recuperação e outra para os casos em que a recuperação é utilizada para corrigir o resultado.
¶ II.12.2.1 Descrição da abordagem matemática top-down recomendada para estimativa da IMA
Esta estimativa é baseada nos seguintes documentos de referência [16, 19, 50, 59, 63, 64, 65 e 67] e parte do princípio de que os dados são oriundos de réplicas de ensaios com amostras fortificadas utilizadas na validação do método ou em dados de controle de qualidade, que são as formas mais comuns de se trabalhar na área de RCA.

¶ II.12.2.1.1 Estimativa do componente da incerteza do Viés para os casos em que não ocorre a correção pela recuperação:
Calcular a incerteza do Viés conforme a fórmula abaixo, o qual faz uso do RQM do Viés, que é a Raiz Quadrada da Média dos quadrados do Viés relativo [67]:

Portanto, temos que o valor do RQM do Viés leva em conta o valor do Viés médio e a sua variação (desvio padrão) [65 e 67].
Quando padrões analíticos certificados e instrumentos calibrados/verificados são usados para preparar as amostras fortificadas, e estes possuem incertezas adequadas, pode-se supor que a incerteza associada ao nível fortificado é insignificante. A Equação então simplifica para:
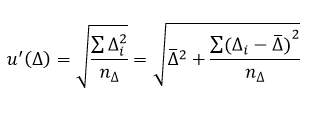
¶ II.12.2.1.2 Estimativa do componente da incerteza do Viés para os casos em que ocorre correção pela recuperação:
Caso o resultado da análise seja corrigido matematicamente para recuperação usando um fator de recuperação, então a incerteza do Viés pode ser calculada usando a seguinte equação, onde DPRRR corresponde ao desvio padrão relativo da recuperação utilizada para corrigir matematicamente o resultado.
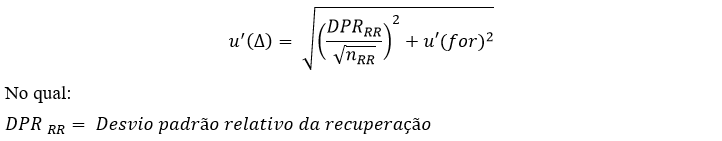
Quando padrões analíticos certificados e instrumentos calibrados/verificados são usados para preparar as amostras fortificadas, e estes possuem incertezas adequadas, pode -se supor que a incerteza associada ao nível fortificado é insignificante. A equação então simplifica para:
¶ II.12.2.1.3 Incerteza da Precisão
O componente da precisão origina do desvio padrão relativo da reprodutibilidade interna (DPRR (%)):
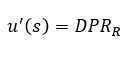
¶ II.12.2.1.4 Estimando a Incerteza combinada
A incerteza combinada para os casos em que não se corrige pela recuperação ao final é definida como:
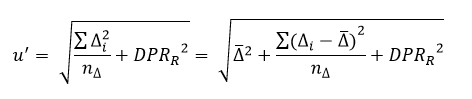
Quando os resultados da análise são corrigidos matematicamente para recuperação usando um fator de recuperação, ao final a incerteza de medição combinada é estimada pela equação:
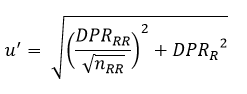
Pela fórmula acima observa-se que caso o valor de nr seja maior ou igual a 9 o impacto do DPRRR da recuperação pode ser desconsiderado.
Recomenda-se que, se possível, os dados relacionados ao Viés e à Recuperação (utilizada para corrigir o resultado), não tenham origem dos mesmos dados do desvio padrão relativo de reprodutibilidade interna (DPRR (%)).
¶ II.12.2.2 Avaliando a significância do Viés
É um requisito geral que as correções sejam aplicadas para todos os erros sistemáticos reconhecidos e significativos, assim é necessário avaliar a significância do erro sistemático (Viés) do método frente a incerteza para definir se o impacto pode ser desconsiderado.
Para verificar se um viés conhecido pode ser desconsiderado, recomenda-se a seguinte abordagem [16]:
- Estimar a incerteza combinada sem considerar o viés relevante;
- Compare a incerteza padrão do viés com a incerteza combinada;
- Quando o valor do viés não for significativo em comparação com a incerteza combinada, a incerteza do viés pode ser desconsiderada no cálculo da IMA do método;
- Quando o viés é significativo em comparação com a incerteza combinada, ele deve:
- Corrigir o resultado pela recuperação, levando em consideração a incerteza da correção (Item II.12.2.1.2);
- Considerar o viés na estimativa da incerteza (Item II.12.2.1.1).
Para verificar a significância da incerteza do viés podem ser aplicados testes de comparação, tais como:
- Teste t para verificar se Recuperação / Veracidade estatisticamente difere de 100% [16 e 63];
- Teste F para comparar a incerteza padrão da reprodutibilidade com a incerteza padrão do viés;
- Comparação direta entre a incerteza padrão do viés e a incerteza padrão da precisão de reprodutibilidade, uma incerteza padrão do viés maior que 2 vezes o valor da incerteza padrão da reprodutibilidade indica que incerteza do viés é significativa. [64].
¶ II.12.2.3 Procedimento top-down considerando outras fontes de incerteza
O procedimento top-down deve avaliar as principais fontes que contribuem para a incerteza final do método, alguns casos observam-se que outras fontes de incerteza, que não somente a precisão e veracidade do método, podem ser identificadas como uma das principais contribuições, dentre elas podemos destacar as seguintes fontes de contribuição:
- Incerteza associada ao modelo da regressão linear (Curva de calibração);
- Incerteza herdada de materiais volumétricos e gravimétricos que impactam no resultado;
- Incerteza dos padrões de referência certificados utilizados no método;
Caso se já identificado que alguma outra fonte, tais como as descritas acima, contribua significativamente para a incerteza do método ela pode ser combinada com a incerteza padrão calculada conforme o item II.12.1.1.
¶ II.12.3 Estimativa da incerteza para os casos de combinação de analitos
Caso não haja uma recomendação específica, se o resultado de uma análise seja reportado como o somatório de analitos (metabólitos, congêneres, definição do LMR, resíduos marcadores…) a incerteza final utilizada deve ser estimada fazendo a combinação das incertezas dos diversos analitos presentes.
¶ II.12.4 Reavaliando a IMA
Periodicamente o laboratório deve reavaliar a IMA do método utilizado na rotina analítica. Essa reavaliação visa verificar se as condições utilizadas para estimar a incerteza permanecem presentes e com o mesmo impacto com relação ao que foi considerado inicialmente.
Uma das formas mais utilizadas para avaliar a incerteza do método se mantem controlada é avaliar os dados de controle de qualidade da rotina analítica, podendo ser utilizadas ferramentas de carta controle e avaliação do histórico dos valores de veracidade (recuperação) e precisão.
Caso essa reavaliação indique que os fatores utilizados no cálculo não se mantiveram constante ao longo do tempo o laboratório deve recalcular e IMA com base nos dados mais recentes e atualizados.
¶ II.12.5 Cálculo da Incerteza Expandida
Normalmente, a incerteza de medição é reportada como a incerteza de medição expandida (𝑈), ou seja, como a incerteza padrão (𝑢) multiplicada por um fator de abrangência 𝑘 = 2, que para uma distribuição normal (Gaussiana) corresponde a uma probabilidade ocorrência de aproximadamente 95%. Entretanto, quando o grau de liberdade efetivo da incerteza padrão calculada for menor que 20, recomenda-se aumentar o fator de abrangência 𝑘 utilizando o fator correspondente na distribuição de t Student.
¶ Parte III - Estudos de Estabilidade
Os procedimentos apresentados objetivam determinar a estabilidade das soluções padrão e dos analitos nas matrizes de origem animal ou vegetal.
A estabilidade do analito, nas mais diversas situações, deve ser determinada de modo a reproduzir as reais condições de armazenamento, manuseio e análise; levando-se em consideração a matriz específica na qual o analito deverá ser pesquisado.
Dados de publicações científicas podem ser utilizados para demonstrar a estabilidade do analito em solução e/ou na matriz, desde que haja similaridade entre as condições estabelecidas no estudo e no laboratório em questão.
Os estudos de estabilidade também podem ser realizados em pareceria por laboratórios da Rede de Laboratórios Agropecuários, e os dados decorrentes poderão ser utilizados para demonstrar a estabilidade do analito nas diversas situações. Para tanto, é necessário que haja similaridade entre as condições do analito nos laboratórios que realizam os estudos.
As estabilidades dos padrões de calibração e padrões internos, em solução devem ser avaliadas.
Os dados podem ser apresentados graficamente, de forma a facilitar a avaliação.
¶ III.1 Estabilidade do Analito na Matriz não Processada
O estudo visa a demonstrar a estabilidade dos analitos nas matrizes no período entre o recebimento da amostra e o início da análise.
O período estudado deve ser superior ao tempo máximo previsto de armazenamento para o início da análise da amostra.
Este estudo somente deve preferencialmente ser realizado com o uso de matrizes naturalmente contaminadas.
É sugerido que o estudo de estabilidade na matriz não processada seja realizado de forma exploratória, conforme demanda do MAPA.
Para tal o laboratório deve reservar amostras naturalmente contaminadas armazenando-as sob temperaturas de congelamento e analisá-las, periodicamente em intervalos de tempo predeterminados.
O analito é considerado estável por um intervalo de tempo, enquanto os resultados dessas análises indicarem igualdade estatística com o primeiro resultado do início do estudo. Para a avaliação do estudo podem ser utilizados testes de comparação média, estimativa do DPR (%) combinados de todas as amostras [33] e testes de significância como Teste t e ANOVA.
¶ III.2 Estabilidade do Analito na Matriz Processada
O estudo visa a demonstrar a estabilidade dos analitos nas matrizes no período entre o processamento da amostra e o início da análise.
A estabilidade do analito na matriz processada deve ser realizada sempre que o laboratório adote a prática na qual a amostra não é analisada imediatamente após o processamento.
O período estudado deve ser superior ao tempo máximo previsto de armazenamento para o início da análise da matriz processada.
Devem-se tomar amostras de matriz branca homogeneizada. O material deve ser dividido em alíquotas. Cada alíquota deve ser fortificada com a substância a ser avaliada.
Armazenar as alíquotas sob as condições nas quais o laboratório as armazena, no período entre o processamento da amostra e o início da análise.
Analisar réplicas das alíquotas armazenadas periodicamente, em intervalos de tempo pré-determinados, e determinar o período de estabilidade dos analitos estudados procedendo a testes, conforme recomendado no Item III.1.
¶ III.3 Estabilidade do Analito no Extrato de Amostra
O estudo visa a demonstrar a estabilidade dos analitos nos extratos no período entre a extração/digestão da amostra e o início da análise instrumental.
A estabilidade do analito no extrato deve ser realizada sempre que o laboratório adote a prática na qual o extrato não é analisado imediatamente após a extração/digestão.
O período estudado deve ser superior à duração da corrida analítica ou ao tempo máximo previsto de armazenamento e corrida analítica.
Os resultados devem ser comparados com aqueles obtidos da análise dos extratos imediatamente após a extração/digestão ou abertura de amostra.
Devem-se tomar extratos de matriz branca. O extrato deve ser adicionado com a substância a ser avaliada em uma concentração que contemple a faixa de trabalho.
Armazenar os extratos sob as condições nas quais o laboratório os armazena, no período entre a extração/digestão da amostra e a análise instrumental.
Analisar os extratos armazenados periodicamente, em intervalos de tempo pré-determinados, e determinar o tempo máximo de estabilidade dos analitos estudados como recomendado no Item III.1.
¶ III.4 Estabilidade das Soluções Padrão
A estabilidade das soluções padrão deve ser demonstrada sempre que não houver alguma referência prévia para o seu prazo de validade.
A estabilidade da solução padrão deve ser estudada nas condições e no período de armazenamento nos quais a solução padrão é armazenada no laboratório.
Recomenda-se comparar a resposta instrumental gerada por uma solução padrão armazenada com a resposta instrumental gerada por uma solução padrão recém preparada.
O estudo deve ser realizado em todos os tipos de soluções padrão, tais como solução padrão estoque e solução padrão de trabalho (e.g., as soluções utilizadas no preparo da curva de calibração e na fortificação das amostras).
Caso o estudo de estabilidade seja feito em uma solução diluída, essa estabilidade pode ser considerada para soluções mais concentradas, desde que ambas sejam armazenadas sob mesmas condições.
O procedimento é o mesmo da seção anterior comparando-se os sinais das soluções padrão obtidos em diferentes períodos de armazenamento com o sinal da solução recém preparada.
¶ Parte IV - A Rotina Analítica e o Controle de Qualidade
Como a validação não garante que o desempenho do método na rotina analítica do laboratório permanecerá o mesmo perpetuamente, faz-se necessário monitorar a rotina analítica para garantir que os resultados continuam válidos no decorrer do tempo, de acordo com o que foi estabelecido durante a validação.
Todos os procedimentos adotados na rotina analítica deverão ser respaldados pelos estudos de validação do procedimento analítico.
A curva de calibração utilizada na rotina deve ser do mesmo tipo e a mesma faixa de trabalho da que foi estudada na validação do método.
No mínimo uma Amostra de Matriz Branca deve estar presente por batelada de amostras,
As ACQ de Matriz Branca Fortificada devem simular níveis de concentração de interesse, para tal fortificações devem ser feitas em nível baixo, médio e alto sendo que o LMR/TMC deve estar representado em um desses níveis ou próximo a eles.
No mínimo uma ACQ de Matriz Branca Fortificada deve estar presente por batelada de amostra. Os níveis de fortificação devem ser periodicamente alternados para que o estudo contemple os três níveis em um curto intervalo de tempo.
Os resultados das ACQ servirão de base para aceitação ou rejeição da batelada analítica.
Para tanto:
- As ACQ devem ser incorporadas em intervalos regulares, dependendo do número total de amostras da batelada.
- Os dados devem, preferencialmente, ser registrados graficamente, com o uso de ferramentas de carta controle, para uma melhor visualização dos resultados.
Os critérios de aceitabilidade dos resultados das ACQs estão estabelecidos nas seções específicas.
O laboratório deve fazer uso, de cartas de controle para mostrar de forma visual e rápida que o sistema de medição analítica está sobre controle estatístico.
Devem ser utilizadas cartas de controle das recuperações obtidas nas ACQ, de modo que seja possível verificar a validade do resultado do lote de análise e demostrar que o método está sob controle através dos valores de precisão e recuperação média dos dados históricos (e.g., últimos 20 dados).
Minimamente deve ser utilizada carta controle de médias (ou valor único) e de modo que seja possível a avaliação pontual do dado quanto aos limites de atenção (e.g., resultado entre duas vezes e três vezes o desvio padrão) e de controle (e.g., resultado acima de 3x o desvio padrão).
Sempre que viável, devem ser utilizadas ferramentas de carta de controle para a identificação de tendências visando identificar antecipadamente possíveis desvios do método.
O laboratório deve definir em documentação interna a forma de avaliar os resultados da carta controle, bem como as ferramentas utilizadas.
Os resultados obtidos nas AQCs podem ser utilizados para estimar continuamente a reprodutibilidade interna, verificando continuamente a precisão e veracidade do método, bem como a sua incerteza.
¶ Parte V - Extensões de Escopo: Inclusão de Novos Analitos, Novas Matrizes e novos valores de Limites Legais, em procedimentos já validados
É frequente a necessidade de estender o escopo de um método previamente validado devido a mudanças na legislação, ou devido à disponibilidade de novas informações, ou a possibilidade de aplicar o método para outros grupos de matrizes. Nestes casos, uma ampliação do escopo deve ser realizada de forma otimizada e analiticamente adequada, idealmente usando um número reduzido de amostras em comparação com uma validação completa.
Alguns exemplos de motivos para ampliar o escopo são:
- Matriz diferente, e.g., uma mudança de músculo bovino para músculo de equino, mudança de fígado suíno para músculo bovino;
- Analito(s) adicional(is);
- Faixa de concentração estendida, i.e., se o LMR for alterado;
- Mudanças no método, i.e., pequenas mudanças na extração/purificação, uma mudança de sistema de cromatografia, ou uma coluna de GC de um novo fornecedor;
Métodos analíticos que são validados de forma completa usando a validação convencional pode muitas vezes ser estendida através de análise de uma quantidade reduzida de experimentos e amostras.
A parte VI deste manual descreve a forma recomendada para realização dessas extensões de escopo para cada área de atuação, com procedimentos objetivos para cada caso e utilizando referências bibliográficas recomendadas.
¶ Parte VI - Aspectos Específicos da Validação de Procedimentos Analíticos para as Diferentes Categorias de Analitos
Nesta parte, trataremos das especificidades que devem ser prioritariamente observadas na validação dos métodos e durante a rotina analítica para os seis grupos de analitos descritos abaixo.
- Resíduos de Medicamentos Veterinários;
- Resíduos de Agrotóxicos (Pesticidas) em Alimentos;
- Contaminantes Inorgânicos;
- Micotoxinas;
- Dioxinas e PCBs-dl (semelhantes a dioxinas);
- Hidrocarbonetos Policíclicos Aromáticos (HPAs).
Todos os critérios descritos nesta seção foram elaborados com base nos principais documentos de referência para as áreas relacionadas. Informações adicionais foram inseridas pela equipe de edição em complemento aos documentos de referência, visando otimizar os procedimentos, torná-los mais claros e adequados à realidade de um laboratório de rotina.
¶ VI.1 Resíduos de Medicamentos Veterinários;
¶ VI.1.1 Validações (Requisitos Mínimos e Critérios de Aceitação)
A validação de procedimentos analíticos para determinação de medicamentos veterinários em matrizes de origem animal deverá ser conduzida observando-se os seguintes parâmetros de desempenho e os seus respectivos critérios de aceitação:
- Seletividade;
- Efeito Matriz;
- Efeito de Matriz Relativo
- Linearidade;
- Recuperação/Veracidade;
- Precisão (Repetitividade);
- Precisão (Reprodutibilidade Intralaboratorial);
- Limite de Decisão (CCα) e Capacidade de Detecção (CCβ);
- Robustez.
A principal referência utilizada é o documento CIR (EU) 2021/808 [33], assim como os seus guias de implementação [21 - 25].
¶ VI.1.1.1 Seletividade
Analisar, sempre que disponível, 20 ou mais amostras brancas de diferentes indivíduos e averiguar a presença de qualquer interferente (pico cromatográfico, sinal, traço de íon) na região de interesse que se espera a eluição do analito.
Quando aplicável, fortificar amostras em branco representativas numa concentração relevante com substâncias que possam interferir na identificação e/ou na quantificação da substância a analisar.
¶ VI.1.1.1.1 Critérios de Aceitabilidade da Seletividade
O sinal interferente deve ser ≤ 30% do sinal na concentração do Menor Nível Calibrado.
¶ VI.1.1.2 Efeito de Matriz
Aplicar os procedimentos de avaliação do Efeito Matriz descritos no Item II.3.
Caso o método faz uso de Curva de Calibração em Solvente (CCS) para quantificar as amostras ele deve demonstrar que o Efeito Matriz não impacta significativamente nos resultados.
Quando se utiliza curva de calibração na matriz fortificada (CCF) ou curva de calibração matrizada (CCM) não é necessário realizar o estudo de efeito de matriz.
¶ VI.1.1.3 Efeito de Matriz Relativo
O efeito de matriz relativo pode ser descrito como a diferença na resposta analítica entre um padrão em solvente e um padrão em extrato de matriz (ex. Um ponto da Curva de Calibração Matrizada - CCM) com correção por um padrão interno.
Realizar a extração de pelo menos 20 amostras brancas diferentes (podem ser utilizados os extratos obtidos no ensaio de seletividade). Após a extração fortificar o extrato no nível de interesse que pode ser o RTM, LMR ou ainda outro valor dentro da faixa de trabalho do método. Analisar o extrato fortificado e comparar com uma solução pura do analito (em solvente) na mesma concentração e injetada em triplicata. Este procedimento deve ser feito para os padrões (analitos de interesse) e para os padrões internos.
Caso o analito em estudo não faça uso de padrão interno o teste de Efeito Matriz Relativo não é obrigatório, porém ele pode ser realizado de forma equivalente ao proposto abaixo.
¶ VI.1.1.3.1 Cálculos
- Calcular o EMR para o padrão (analito) em cada extrato:

- Calcular o EMR para o padrão interno em cada extrato:

- Calcular o EMR normalizado:

- Calcular o coeficiente de variação para o EMR padronizado (CVEMR):

¶ VI.1.1.3.2 Critérios de aceitação
O coeficiente de variação, CVEMR (%) deve ser menor ou igual a 20%.
¶ VI.1.1.4 Linearidade
Construir uma curva de calibração com no mínimo cinco níveis de concentração, incluindo o “zero”. O intervalo da curva de calibração deve ser construído de tal forma que os níveis avaliados nos estudos de precisão/exatidão estejam dentro da faixa de calibração.
A linearidade deve ser avaliada pelo coeficiente de correlação ou determinação.
Sugere-se fazer uma inspeção visual do gráfico gerado pela regressão linear.
A curva de calibração não deve ser forçada a passar pela origem.
Calcular o coeficiente de correlação ou determinação.
¶ VI.1.1.4.1 Critérios de Aceitabilidade
Para que o modelo seja considerado satisfatório o valor de Coeficiente de Determinação (r2) deve ser maior ou igual a 0,95 (r2 ≥ 0,95), ou seja, o valor do Coeficiente de Correlação (r) deve ser maior ou igual a 0,975 (r ≥ 0,975).
¶ VI.1.1.5 Precisão (Repetitividade e Reprodutibilidade Intralaboratorial)
¶ VI.1.1.5.1 Repetitividade
O presente parâmetro é avaliado por meio da análise de amostras brancas fortificadas. A fortificação das amostras brancas deve ser realizada conforme orientações abaixo:
- Para resíduos de drogas veterinárias com LMR definido (substâncias autorizadas): preparar e analisar, no mínimo, seis replicatas de amostra branca fortificadas em três níveis de concentração (0,1 LMR – 1,0 LMR – 1,5 LMR)(1). Repetir este procedimento pelo menos mais duas vezes;
- Para resíduos de drogas veterinárias de uso proibido ou não autorizado com RTM definido: preparar e analisar, no mínimo, seis replicatas de amostra branca fortificadas em três níveis de concentração (0,5 RTM – 1,0 RTM – 1,5 RTM)(2). Repetir este procedimento pelo menos mais duas vezes;
- Para resíduos de drogas veterinárias de uso não autorizado sem RTM definido: preparar e analisar, no mínimo, seis replicatas de amostra branca fortificadas em três níveis de concentração (1,0 TMC – 2,0 TMC - 3,0 TMC). Repetir este procedimento pelo menos mais duas vezes;
(1) Se, para uma substância farmacologicamente ativa específica, a validação de uma concentração de 0,1 vezes o LMR não for razoavelmente possível, a concentração de 0,1 vezes o LMR pode ser substituída pela concentração mais baixa entre 0,1 e 0,5 vezes o LMR, que seja razoável alcançar;
(2) Se, para uma substância farmacologicamente ativa não autorizada, a validação de uma concentração de 0,5 vezes o RTM não for razoavelmente possível, a concentração de 0,5 vezes o RTM pode ser substituída pela concentração mais baixa entre 0,5 e 1,0 vezes o RTM, que seja razoável alcançar;
¶ VI.1.1.5.1.1 Cálculos
Calcular o coeficiente de variação da repetibilidade (CVrepe), equivalente ao DPRr (%), conforme descrito no Item II.4.4.
Realizar, se necessário, avaliação de presença de outliers utilizando testes estatísticos tais como os testes de Grubbs.
¶ VI.1.1.5.1.2 Critérios de aceitação
Avaliar a aceitabilidade do valor de CVrepro conforme descrito na Tabela 3 abaixo.
Tabela 3: Critérios de aceitação do coeficiente de variação (CV) para a repetibilidade.
|
Faixas de Concentração (μg/kg ou μg/L) |
CV (%) |
|
Conc < 10 |
20,0 |
|
10 ≤ Conc ≤ 120 |
16,7 |
|
120 < Conc ≤ 1000 |
14,7 |
|
Conc >1000 |
10,7 |
¶ VI.1.1.5.2 Reprodutibilidade intralaboratorial
Proceder conforme descrito no item VI.1.6.1 atentando para o fato de repetir o experimento pelo menos mais duas vezes em dia(s) diferente(s) e se possível variando o analista, equipamentos etc.
¶ VI.1.1.5.2.1 Cálculos
Calcular o coeficiente de variação da reprodutibilidade interna (CVrepro), equivalente ao DPRR (%), conforme descrito no Item II.4.4.
Realizar, se necessário, avaliação de presença de outliers utilizando testes estatísticos tais como testes de Grubbs.
¶ VI.1.1.5.2.2 Critérios de aceitação
Avaliar a aceitabilidade do valor de CVrepro conforme descrito na Tabela 4 abaixo.
Tabela 4: Critérios de aceitação do coeficiente de variação (CV) para a reprodutibilidade intralaboratorial
|
Faixas de Concentração (μg/kg ou μg/L) |
CV (%) |
|
Conc < 10 |
30 |
|
10 ≤ Conc ≤ 120 |
25 |
|
120 < Conc ≤ 1000 |
22 |
|
Conc >1000 |
16 |
¶ VI.1.1.6 Veracidade/Recuperação
Com os dados obtidos no ensaio de reprodutibilidade intralaboratorial calcular a veracidade / recuperação média, por nível de fortificação.
¶ VI.1.1.6.1 Critérios de aceitação
Avaliar a aceitabilidade do valor de veracidade / recuperação conforme descrito na Tabela 5 abaixo
Tabela 5: Critérios de aceitação da veracidade/recuperação
|
Faixas de Concentração (μg/kg ou μg/L) |
Recuperação (%) |
|
Conc ≤ 1 |
50 a 120 |
|
1 < Conc < 10 |
70 a 120 |
|
Conc ≥ 10 |
80 a 120 |
¶ VI.1.1.7 Incerteza padrão combinada
Tendo em vista que para análises de resíduos de drogas veterinárias a incerteza de medição padrão combinada é considerada no valor CCα (limite de decisão para a confirmação), a estimativa da incerteza padrão combinada tem como objetivo gerar subsídios para os cálculos de CCα e CCβ (Capacidade de Detecção para triagem).
Estimar a Incerteza padrão combinada conforme descrito no Item II.12.
¶ VI.1.1.8 Limite de Decisão para Confirmação (CCα) e Capacidade de Detecção para triagem (CCβ)
¶ VI.1.1.8.1 Limite de Decisão para Confirmação (CCα)
Deve determinar-se o CCα para os métodos de confirmação.
- Para as substâncias farmacologicamente ativas não autorizadas no Brasil, não autorizadas para a espécie, ou proibidas, o CCα deve ser calculado utilizando uma das seguintes opções:
- a) Opção 1: através do procedimento da curva de calibração de acordo com a norma ISO 11843-1:1997 [44]. Para tanto, deve-se utilizar material em branco, fortificado no e acima do RTM ou TMC em passos equidistantes. Analisar as amostras. Após a identificação, representar graficamente o sinal quando possível, ou a concentração recalculada em função da concentração adicionada. O limite de decisão é igual à concentração correspondente à ordenada na origem mais 2,33 vezes o desvio-padrão da reprodutibilidade intralaboratorial. Este método é aplicável apenas aos ensaios quantitativos. Os limites de decisão obtidos com esta abordagem devem ser verificados analisando a matriz em branco fortificada no limite de decisão calculado.
- b) Opção 2: CCα = TMC + k (unilateral, 99%) × incerteza de medição-padrão (combinada) no TMC. Para as substâncias farmacologicamente ativas não autorizadas ou proibidas, dependendo da experiência de validação (e dos respetivos graus de liberdade), a distribuição-t pode ser razoavelmente aplicada, ou ainda, pode ser tomada como base a distribuição de Gauss (unilateral, n=∞), utilizando um fator k de 2,33.
2. Para as substâncias autorizadas, o CCα deve ser calculado utilizando uma das seguintes opções:
i) No caso das substâncias autorizadas em combinações matriz/espécie para as quais tenha sido fixado um LMR:
- a) Opção 1: através do procedimento da curva de calibração de acordo com a norma ISO 11843-1:1997. Para tanto, deve-se utilizar material em branco, fortificado no e acima do LMR em passos equidistantes. Analisar as amostras. Após a identificação, representar graficamente o sinal quando possível, ou a concentração recalculada em função da concentração adicionada. O limite de decisão (α = 5%) é igual à concentração correspondente ao LMR mais 1,64 vezes o desvio-padrão da reprodutibilidade intralaboratorial no nível permitido. Os limites de decisão obtidos com esta abordagem devem ser verificados analisando a matriz em branco fortificada no limite de decisão calculado.
- b) Opção 2: CCα = LMR + k (unilateral, 95%) × incerteza de medição-padrão (combinada) no LMR.
Para as substâncias autorizadas, dependendo da experiência de validação (e dos respetivos graus de liberdade), a distribuição-t pode ser razoavelmente aplicada, ou ainda, pode ser tomada como base a distribuição de Gauss (unilateral, n=∞), utilizando um fator k de 1,64.
No caso das substâncias farmacologicamente ativas para as quais está estabelecido um LMR para a soma de diferentes substâncias, o CCα da substância com a concentração mais elevada na amostra deve ser utilizado como CCα para avaliar a soma das substâncias na amostra medida.
¶ VI.1.1.8.2 Capacidade de Detecção para triagem (CCβ)
Deve-se determinar a CCβ apenas para os métodos de triagem (qualitativos).
1. Para as substâncias farmacologicamente ativas não autorizadas ou proibidas, deve ser assegurado um erro do tipo β máximo de 5%. A CCβ deve ser calculada utilizando uma das seguintes opções:
- a) Opção 1: através do procedimento da curva de calibração de acordo com a norma ISO 11843-1:1997. Para tanto, deve ser utilizado material em branco representativo, fortificado ao RTM e abaixo ou, se não tiver sido estabelecido nenhum RTM, a fortificação deve ser feita em valores próximos ao da concentração-alvo para a triagem (STC — screening target concentration) em passos equidistantes [22]. Analisar as amostras. Representar graficamente o sinal em função da concentração adicionada. A capacidade de detecção é igual à concentração correspondente à STC mais 1,64 vezes o desvio-padrão da reprodutibilidade intralaboratorial do teor médio medido no nível do STC. A extrapolação muito abaixo do nível mais baixo de fortificação (< 50% do nível mais baixo de fortificação) deve ser confirmada por dados experimentais na fase de validação.
- b) Opção 2: investigação de material em branco fortificado a níveis de concentração iguais ou superiores à STC. Para cada nível de concentração, devem analisar-se 20 amostras em branco fortificadas, a fim de garantir uma base fiável para esta determinação. A capacidade de detecção do método é igual ao nível de concentração no qual apenas se obtém ≤ 5% de falsos resultados conformes.
- c) Opção 3: CCβ = STC + k (unilateral, 95%) × incerteza de medição-padrão (combinada) no nível do STC ou acima.
Para as substâncias farmacologicamente ativas não autorizadas ou proibidas, dependendo da experiência de validação (e dos respetivos graus de liberdade), a distribuição-t pode ser razoavelmente aplicada, ou ainda, pode ser tomada como base a distribuição de Gauss (unilateral, n=∞), utilizando um fator k de 1,64.
1. Para as substâncias autorizadas, deve ser assegurado um erro máximo β de 5%. A CCβ deve ser calculada do seguinte modo:
- a) Opção 1: através do procedimento da curva de calibração de acordo com a norma ISO 11843-1:1997. Para tanto, deve ser utilizado material em branco representativo, fortificado ao nível e abaixo do limite permitido, a partir da STC em passos equidistantes [22]. Analisar as amostras e identificar o(s) analito(s). Calcular o desvio-padrão do teor médio medido no nível do STC. A capacidade de detecção é igual à concentração correspondente ao nível do STC mais 1,64 vezes o desvio-padrão da reprodutibilidade intralaboratorial do teor médio medido na STC.
- b) Opção 2: através da investigação de material em branco fortificado a níveis de concentração inferiores ao limite permitido. Para cada nível de concentração, devem analisar-se 20 amostras em branco fortificadas, a fim de garantir uma base fiável para esta determinação. A capacidade de detecção do método é igual ao nível de concentração no qual apenas se obtém ≤ 5% de falsos resultados conformes.
- c) Opção 3: CCβ = STC + k (unilateral, 95%) × incerteza de medição-padrão (combinada) no nível do STC ou acima.
Para as substâncias autorizadas, dependendo da experiência de validação (e dos respetivos graus de liberdade), a distribuição-t pode ser razoavelmente aplicada ou, se se tomar como base a distribuição de Gauss (unilateral, n=∞), deve ser utilizado um fator k de 1,64 (mediante seja a utilização em cascata, seja a utilização regular do LMR).
No caso das substâncias farmacologicamente ativas para as quais está estabelecido um LMR para a soma de diferentes substâncias, a CCβ da substância com a concentração mais elevada na amostra deve ser utilizada como CCβ para avaliar a soma das substâncias na amostra medida.
¶ VI.1.1.8.3 Critérios de Aceitação do CCα e CCβ
No caso de substâncias proibidas que possuem RTM, os valores de CCα e CCβ devem ser inferiores ao valor de Referência para Tomada de Medida – RTM.
¶ VI.1.1.9 Robustez
Um estudo de robustez deve ser realizado demonstrando a estabilidade do método sob diferentes condições, diferentes fabricantes de insumos, colunas cromatográficas dentre outras variações, conforme descrito no Item II.8.
Entretanto, a avaliação da robustez também pode ser demonstrada pelo acompanhamento dos resultados das amostras de controle de qualidade ao longo da rotina analítica, de modo que seja demonstrado que os valores de reprodutibilidade (DPRR) e Veracidade (recuperação média) estão se mantendo adequados ao longo do tempo, do mesmo modo que é realizada a verificação concorrente de desempenho do método.
A adequação dos valores monitorados demonstra a robustez adequada do método, considerando que ao longo da rotina analítica ocorre diversas mudanças, tais como diferentes lotes de reagentes e insumos, diferentes equipamentos quantitativos e qualitativos utilizados na rotina (balanças, pipetas, micropipetas, balões volumétricos, agitadores, centrífugas), simulando de forma mais realista às modificações que podem ocorrer.
¶ VI.1.2 Estudos de Estabilidade
A estabilidade dos analitos utilizados nas análises deve ser demonstrada, simulando as condições as quais o laboratório submete as amostras/padrões.
Os estudos de estabilidade podem ser realizados ao longo dos ensaios de rotina.
¶ VI.1.2.1 Critério de Aceitação
Os analitos serão considerados estáveis quando for observada uma degradação máxima de até 15%, em relação a uma referência recém preparada.
¶ VI.1.3 Extensões de Escopo: Inclusão de Novos Analitos, Novas Matrizes e novos valores de LMR ou RTM
Se um método for alterado ou ampliado, a aplicação de um esquema de validação reduzido é, em geral, possível. No entanto, o tipo e o número de variações a serem validadas em uma validação reduzida deve sempre basear-se em conhecimentos especializados e experiências anteriores [24].
Geralmente, a extensão do método para novos analitos só é possível para analitos que podem ser incluídos no método analítico sem divergência da descrição do método. Se esses requisitos são cumpridos, a aplicação de um esquema de extensão de escopo para novos analitos pode ser utilizada.
A extensão de escopo pode ser realizada de duas formas: 1 - Utilizando os dados de controle de qualidade na rotina analítica, camada de verificação concorrente (on going) de desempenho do método, e 2- Realizar a chamada extensão do escopo por validação reduzida.
Em todos os casos de extensão de escopo é necessário que, após a extensão do escopo seja realizado o monitoramento do desempenho de método através do controle de qualidade, de forma a complementar a validação do método com dados de verificação contínua na rotina analítica.
¶ VI.1.3.1 Extensão de escopo de forma concorrente (on going)
Para essa extensão de escopo o analista deve utilizar os resultados obtidos pelas Amostras de controle de qualidade (ACQ), que deve conter no mínimo 3 níveis de concentração e uma amostra branca por dia / lote de análise, sendo que ele deve combinar os dados de pelo menos 5 dias / lotes de análise.
Recomenda-se que seja utilizado amostras brancas de diferentes indivíduos para avaliar de forma equivalente à validação completa.
Realizar a fortificação das ACQ de acordo com as orientações descritas em VI.1.6.1, utilizando os mesmos níveis recomendados para validação direta.
¶ VI.1.3.2 Extensão de escopo por validação reduzida
Os níveis de fortificação para uma validação reduzida são os mesmos descritos no item VI.1.6.1. São necessários seis lotes de matriz branca (por exemplo, seis amostras diferentes de músculo bovino).
Repetir o processo sob condições de repetibilidade, até que se obtenham seis análises por nível, incluindo o nível zero (amostra branca).
¶ VI.1.3.3 Estimando os parâmetros de validação da extensão de escopo
Verificar a linearidade do método estendido conforme recomenda Item VI.1.5.
Calcular a recuperação / veracidade média e o coeficiente de variação por nível fortificado, conforme recomenda os itens VI.1.6 e VI.1.7.
Calcular os valores de CCα e CCβ, conforme a aplicabilidade, com os dados obtidos, conforme descrito nos Item VI.1.9, quando aplicável. Recomenda-se utilizar a opção de estimar o CCα utilizando a incerteza-padrão combinada no LMT, ou TMC, conforme for o caso.
Caso a Extensão de escopo tenha sido realizada em condições de repetibilidade recomenda-se multiplicar a precisão (desvio padrão) da repetibilidade por um fator de 1,5 [24].
Os valores de veracidade / recuperação média e de coeficiente de variação obtidos devem estar dentro dos intervalos aceitáveis utilizados na validação do método.
Avaliar a Seletividade Efeito Matriz Relativo utilizando as amostras brancas, conforme recomenda nos itens VI.1.2 e VI.1.4.
A linearidade deve atendar aos critérios especificados no item VI.1.5.1 Devem ser avaliados a Seletividade e o Efeito Matriz Relativo, conforme descrito nos Item VI.1.2 e VI.1.4 respectivamente.
Estimar a Incerteza-padrão combinada conforme Item II.12, que será utilizada no cálculo do CCα do escopo estendido.
¶ VI.1.4 Curvas de Calibração e Amostras de Controle de Qualidade (ACQs) na Rotina Analítica
Todos os procedimentos adotados na rotina analítica deverão ser respaldados pelos estudos de validação do método.
A curva de calibração utilizada na rotina deve conter pelo menos cinco níveis (incluindo o zero), devendo esta ter a mesma faixa de trabalho estudada na validação do método.
Para avaliar veracidade do lote de análise ACQ devem estar presentes pelo menos no nível de concentração do LMR ou TMC.
Os valores de recuperação / veracidade obtidos pelos ACQs devem estar dentro dos limites estabelecidos no Item VI.1.1.6.
Ferramenta de carta de controle de média e/ou de desvio padrão deve estar presentes e deve ser utilizadas para avaliar o desempenho dos resultados de controle de qualidade do lote, assim como deve ser utilizada apara avaliar possíveis tendências e para demonstrar que o método está sob controle quanto à precisão e veracidade / recuperação ao longo da rotina analítica.
Para maiores detalhes com relação a construção e avaliação da carta controle verificar o Item IV deste manual.
¶ VI.2 Resíduos de Agrotóxicos (Pesticidas) em Alimentos;
A análise de resíduos de agrotóxicos em alimentos tem como principal documentação guia o documento SANTE [19], as orientações contidas neste documento destinam-se aos laboratórios envolvidos no controle oficial de resíduos de agrotóxicos em gêneros alimentícios e alimentos para animais em toda a União Europeia (UE). O documento descreve os requisitos de validação de métodos e de controle de qualidade analítico para subsidiar a validade dos dados no âmbito dos controles oficiais de resíduos de agrotóxicos, e utilizados para verificar a conformidade com níveis máximos de resíduos (LMR), ações de fiscalização ou avaliação da exposição do consumidor.
Além do documento SANTE, dois documentos Guias publicados pela Comissão Codex Alimentarius CXG 90-2017 [13] e o CXG 40-1993 [9] que tratam de boas práticas laboratoriais e critérios de desempenho para métodos de análise de resíduos de agrotóxicos em alimentos, são referências internacionais amplamente utilizadas, os conceitos apresentados nos documentos complementam diversas informações presentes no documento SANTE e ao mesmo tempo define procedimentos e critérios de desempenho mais detalhados para validação de métodos e manutenção na rotina analítica.
Nesta seção, além da conciliação dos três documentos guias citados acima, adiciona-se a vasta experiência prática dos LFDAs na aplicação destes em atendimento à rotina de análises oficiais do MAPA.
¶ VI.2.1 Processamento da amostra
O processamento das amostras é a etapa inicial de toda análise de resíduos de agrotóxicos em alimentos, sendo influenciada pelo modo a realizá-lo, devendo ser de forma representativa e em acordo com a forma que se estabelece os valores de LMR. O laboratório deve definir protocolos internos descrevendo a etapa do processamento das amostras, seguindo as orientações dos documentos Regulamento (UE) 752/2014 da Comissão [32] e o documento CAC/GL 41-2010 [8] que especificam quais partes dos alimentos devem ser processados, conforme a particularidade da matriz e outras informações correlacionadas.
¶ VI.2.2 Matrizes Representativas
Para melhor definir o agrupamento dos tipos de matrizes que requerem análise de resíduos de agrotóxicos, foi estabelecida uma classificação com base em características do produto, como umidade (teor de água), acidez, presença de lipídios e outras. Esse processo resultou na definição de grupos de matrizes conforme detalhado na Tabela 6, a qual foi elaborada com base no Anexo A do documento SANTE [19].
Tabela 6: Grupos de produtos e as espécies representativas.
|
Grupo de produto |
Propriedades comuns |
Classe do produto |
Espécies representativas |
|
Produtos de Origem Vegetal |
|||
|
I. |
Alto teor de água |
Folhas verdes e brássicas |
Espinafre, alface, brócolis, repolho, couve |
|
Legumes verdes |
Feijão verde |
||
|
Pomáceas |
Maçã, pera |
||
|
Frutas de Caroço |
Pêssego, cereja, manga |
||
|
Bagas |
Uva |
||
|
Frutos pequenos |
Morango |
||
|
Frutos pequenos |
Morango |
||
|
Vegetais em frutos |
Tomate, pimentão, melão, kiwi, abacaxi, pepino |
||
|
Cogumelos |
Cogumelos |
||
|
Legumes |
Abóbora, abobrinha, chuchu |
||
|
Vegetais de bulbo |
Cebola |
||
|
Vegetais em raízes |
Batata, cenoura, salsão |
||
|
II. |
Alto teor ácido |
Frutas cítricas |
Laranja, limão e mexerica |
|
III. |
Alto teor de açúcar |
Frutas desidratadas |
Passas, tâmaras, geleia |
|
IV. |
Alto teor de óleo ou gordura |
Sementes oleaginosas |
Abacate, semente de girassol, amendoim |
|
Castanhas |
Noz, noz pecam, pistache, soja |
||
|
V. |
Materiais secos |
Cereais |
Trigo, arroz, milho, feijão |
|
Produtos de cereais |
Farelo de trigo, farinha de trigo |
||
|
VI. |
Produtos que requerem testes individuais |
- |
Alho, café torrado (grão ou moído), café verde (grão), chá, folhas secas, gengibre, lúpulo, temperos |
|
Produtos de Origem Animal |
|||
|
VII. |
Carnes |
Músculo bovino, suíno, equino, de pescado e de ave |
|
|
Grupo de produto |
Propriedades comuns |
Classe do produto |
Espécies representativas |
|
VIII. |
Miúdos |
Fígado, rim |
|
|
IX. |
Gordura |
Gordura de carne |
|
|
X. |
Leite |
Leite de vaca |
|
|
XI. |
Ovo |
Ovos de galinha |
|
|
XII. |
Mel |
Mel, geleia real, cera |
|
O procedimento de validação do método deve ser realizado de forma completa para pelo menos uma matriz representativa para cada grupo de matriz validado.
Quando um método for implementado na rotina para uma variedade de matrizes de um mesmo grupo, dados complementares deverão ser coletados de forma a confirmar a validade do método para aquelas matrizes específicas do grupo, esses dados podem ser avaliados em paralelo à rotina analítica, com a chamada verificação concorrente (do termo em inglês on going). Ver maiores detalhes para esse procedimento no Item VI.2.8.
¶ VI.2.3 Validações (Requisitos Mínimos e Critérios de Aceitação)
A validação de procedimentos analíticos para determinação de agrotóxicos em matrizes de origem vegetal, animal e rações, deverá ser conduzida atendendo aos critérios mínimos dos seguintes parâmetros de desempenho:
- Seletividade (interferentes);
- Efeito Matriz;
- Linearidade;
- Recuperação/Veracidade;
- Precisão (Repetitividade e Reprodutibilidade intralaboratorial);
- Limite de Quantificação;
- Razão de íons;
- Robustez (opcional);
- Incerteza de Medição.
¶ VI.2.3.1 Seletividade (Especificidade)
Analisar no mínimo três Amostras Brancas, verificar a ocorrência de interferentes (sinais e/ou picos no tempo de retenção em que se prevê a eluição do analito).
Caso seja identificado algum interferente nas amostras Brancas recomenda-se que seja analisado uma amostra de Branco de Reagentes para identificar a origem do interferente.
¶ VI.2.3.1.1 Critérios de Aceitabilidade
O sinal médio obtido nas amostras brancas deve ser ≤ 30% do sinal na concentração do Limite de Quantificação (ou do menor nível calibrado).
¶ VI.2.3.2 Razão de Íons
Realizar a verificação da razão de íons conforme recomenda o Item II.7 com os dados da validação do método.
¶ VI.2.3.2.1 Critérios de Aceitabilidade
A variação da razão de íons não deve ultrapassar o valor de ±30% para ser considerado adequado.
Analitos com variação acima de 30% podem ser aceitos, porém um cuidado maior deve ser tomado na rotina analítica, pois para esses analitos a confirmação da presença na amostra provavelmente será prejudicada.
¶ VI.2.3.3 Efeito Matriz (EM)
Aplicar os procedimentos de avaliação do Efeito Matriz conforme descrito no Item II.3 deste manual.
Caso a ocorrência de EM já seja conhecida a necessidade de se trabalhar com CCM ou CCF já tenha sido identificada, não há a necessidade de realizar o estudo de EM.
O estudo de EM deve obrigatoriamente ser realizado nos casos em que for utilizada a CCS para quantificar as amostras.
¶ VI.2.3.3.1 Critérios de Aceitabilidade
A ocorrência de Efeito Matriz fora da faixa de ± 20% indica que o efeito de matriz precisa ser considerado ao preparar a curva de calibração, logo, curvas de calibração em solvente (CCS) não poderá ser utilizada.
No caso de presença de efeito matriz fora do intervalo de ± 20%, é necessário o uso da CCM ou da CCF para quantificar as amostras.
¶ VI.2.3.4 Linearidade
Construir uma curva de calibração com no mínimo cinco níveis de concentração, compreendendo os valores de LQ e 10 vezes o LQ, preparar cada nível de calibração pelo menos em triplicata (três vezes cada nível de calibração);
Recomenda-se utilizar a regressão MMQP, 1/v, 1/x, 1/y, 1/x2 ou 1/y2, como forma de se adequar à heteroscedasticidade dos dados, maiores detalhes verificar o Item II.2 do manual.
Regressão de 2° grau (quadrática), deve ser evitada, entretanto ela pode ser utilizada para descrever analitos com comportamento específico. Regressões de graus mais elevados não podem ser utilizadas.
Calcular o coeficiente de correlação (r) ou o coeficiente de determinação (R2) e o Desvio Relativo de cada nível da curva de calibração (ver Item II.2.2).
¶ VI.2.3.4.1 Critérios de Aceitabilidade
Os desvios relativos por nível de calibração não devem ser maiores que ± 20%, ou seja, o valor de Recuperação da concentração retrocalculada (Rcr (%)) por nível de calibração deve ficar entre 80 e 120%.
O valor do coeficiente de determinação R2 deve ser maior ou igual a 0,96 (R2 ≥ 0,96), ou seja, o valor do coeficiente de correlação, r, deve ser maior ou igual a 0,98 (r ≥ 0,98).
¶ VI.2.3.5 Veracidade/Recuperação e Precisão (Repetitividade e Reprodutibilidade Parcial)
Realizar a fortificação de, no mínimo, cinco replicatas de amostras em pelo menos dois níveis de concentração: Um nível baixo que corresponda ao Limite de Quantificação (LQ) e em um nível mais alto.
Para a determinação da reprodutibilidade intralaboratorial (precisão intermediária), repetir os procedimentos acima no mínimo em mais duas ocasiões (em condições de reprodutibilidade).
Calcular a recuperação / veracidade média e os valores de desvio padrão relativo de repetibilidade (DPRr) e de reprodutibilidade intralaboratorial (DPRR) por nível de concentração, conforme recomendado no Item II.4.4.
O valor de Desvio Padrão Relativo de reprodutibilidade intralaboratorial também pode ser obtido com os dados de controle de qualidade do método (Verificação concorrente - on going), conforme descrito no Item VI.2.8.
¶ VI.2.3.5.1 Critérios de Aceitabilidade
O método deve obter um valor de Veracidade (Recuperação) média, em cada nível de fortificação de 70% a 120%.
O valor de Desvio Padrão Relativo de repetibilidade (DPRr) deve ser inferior ou igual a 20%.
Nos casos em que a Veracidade média situar fora da faixa estabelecida, porém o valor possui uma boa precisão (i.e., possui DPRr ≤ 20%) e a razão para isto é bem estabelecida (i.e., analitos que tem histórico de apresentar baixa recuperação, tais como os de alta polaridade e os de alta lipofilicidade), valores de Veracidade fora da faixa de 70 a 120% pode ser aceitável, entretanto a veracidade média não pode ser abaixo de 30% ou acima de 140% independente da precisão.
Caso o valor de recuperação fique fora da faixa de 70 a 120% duas abordagens podem ser praticadas:
- Fazer uma correção matemática dos resultados pelo valor de recuperação estabelecido;
- Trabalhar com CCF (ou adição de padrão) ou com uso de padrão interno isotopicamente marcado (que deve ser adicionado antes do início da extração);
Para o valor de Desvio Padrão Relativo de reprodutibilidade interna DPRR obtido na validação do método recomenda-se utilizar os limites aceitáveis indicados no documento CAC CGX/40-1993 [9] no qual utiliza-se dos limites de DPR estimados pela equação de Horwitz [37] a seguir:
DPR = 2 (1-0,5 log C)
Em que, C corresponde à concentração da amostra expressa em termos de fração mássica (g/g)
Na Tabela 7 abaixo estão alguns exemplos de estimativas de limite para DPRR calculados.
Tabela 7: Valores de limite de DPRR estimado para as concentrações apresentadas.
|
Concentração (mg/kg) |
Limite para DPRR (%) |
|
0,01 |
32 |
|
0,02 |
28,8 |
|
0,05 |
25,1 |
|
0,1 |
22,6 |
Para concentrações abaixo de 0,01 mg/kg, o limite para DPRR deve ser tão baixo quanto possível, para esses níveis de concentração recomenda-se utilizar o valor correspondente a concentração 0,01 mg/kg (32%).
Levando em conta que o valor máximo aceito para a incerteza expandida do método é 50%, valores de DPRR acima de 25% podem impedir que a incerteza fique menor que 50% o que limitará a adequação do método para o analito em questão.
¶ VI.2.3.6 Limites de Quantificação
O limite de quantificação do procedimento analítico é definido como o nível de concentração mais baixo no qual foi demonstrado que os critérios de veracidade e precisão foram atendidos.
¶ VI.2.3.7 Robustez
A avaliação da robustez durante a validação do método não é obrigatória, porém ela pode ser realizada conforme descrito no Item II.8.
Para a área de resíduos de agrotóxicos a avaliação da robustez deve ser demonstrada pelo acompanhamento dos resultados das amostras de controle de qualidade ao longo da rotina analítica, de modo que seja demonstrado que os valores de reprodutibilidade (DPRR) e Veracidade (recuperação média) estão se mantendo adequados ao longo do tempo, do mesmo modo que é realizada a verificação concorrente de desempenho do método.
A adequação dos valores monitorados demonstra a robustez adequada do método, considerando que ao longo da rotina analítica ocorre diversas mudanças, tais como diferentes lotes de reagentes e insumos, diferentes equipamentos quantitativos e qualitativos utilizados na rotina (balanças, pipetas, micropipetas, balões volumétricos, agitadores, centrífugas), simulando de forma mais realista às modificações que podem ocorrer.
¶ VI.2.4 Incerteza de Medição
A incerteza de medição deve ser estimada segundo procedimento estabelecido no Item II.12.
¶ VI.2.4.1 Critérios de Aceitabilidade
A incerteza de medição expandida (U), relativa, deverá ser inferior ou igual a 50% para o nível de confiança de 95%.
¶ VI.2.5 Estudos de Estabilidade (Orientação Geral)
Amostras devem ser armazenadas por um período curto, que não afete a estabilidade dos agrotóxicos [17], devendo ser mantidas em temperaturas próximas à -20°C, temperaturas dessa magnitude promovem uma estabilidade adequada à amostra na medida que a degradação dos agrotóxicos devido à atividade enzimática é extremamente baixa [9].
Caso o laboratório analise as amostras em curto intervalo de tempo (i.e., entorno de 30 dias), mantendo as condições de armazenamento adequadas para cada tipo de matriz, a realização do estudo de estabilidade não é mandatória.
Os estudos de estabilidade podem ser realizados de forma concorrente à rotina analítica e deve simular as condições nas quais o laboratório submete as amostras/padrões (e.g. se o laboratório armazena as amostras processadas e congeladas por até 90 dias ele deve provar a estabilidade dos analitos nas amostras nas mesmas condições).
¶ VI.2.6 Extensões de Escopo (Novos Analitos)
Quando se desejar incluir novos analitos a um método previamente validado, o laboratório deve realizar o procedimento de validação de método de forma integral para o analito em questão.
De maneira alternativa, a validação de novo analito pode ser realizada pela verificação concorrente (on going) conforme definido no Item VI.2.8.
¶ VI.2.7 Controle de qualidade do lote de análise
¶ VI.2.7.1 Calibrações Analíticas e Recuperação (Rotina Analítica)
Todos os procedimentos adotados na rotina analítica deverão ser respaldados pelos estudos de validação do método.
Todo resultado acima do LQ deve ser quantificado com o uso de curva de calibração validada e a veracidade (recuperação) do analito quantificado deve ser verificada nas amostras de controle de qualidade do lote.
A matriz selecionada para a preparação da curva de calibração e das amostras de controle de qualidade deve ser do mesmo grupo de matriz correspondente às amostras quantificadas.
Caso os resultados quantificados estejam próximo ou acima do LMR, recomenda-se que a curva de calibração (CCM ou CCF) sejam preparados na mesma matriz da amostra, esse procedimento deve ser realizado principalmente para os analitos que possuem efeito matriz mais pronunciado, tais como os analitos que eluem no início da cromatografia líquida.
A resposta do detector dos analitos nas amostras analisadas deve estar dentro da faixa da curva de calibração. Caso a concentração calculada na amostra esteja 20% ou mais acima do maior nível de calibração é necessário que a amostra seja diluída (normalmente o extrato é diluído) e reinjetado.
Ressalta-se que caso a quantificação tenha sido realizada em CCM ou CCF a diluição da amostra deve ser feita com uma amostra branca de modo que seja mantido a mesma proporção matriz/extrato que a amostra.
¶ VI.2.7.1. a) Monitoramento das Curvas de Calibração
O monitoramento das curvas de calibração deve ser realizado para todos os analitos do escopo que estão sendo analisados no lote.
¶ VI.2.7.1. b) Monitoramento dos valores de Recuperação (Veracidade)
A verificação da recuperação, na rotina, é realizada conforme a Tabela 8 abaixo, somado dos analitos quantificados nas amostras do lote.
Tabela 8: Frequência mínima de determinação da recuperação na rotina analítica
|
Analitos Obrigatórios |
Todos os Outros Analitos do Escopo |
|
Pelo menos 10% dos Analitos do escopo (pelo menos 5 em cada lote de análises). |
Dentro de um programa contínuo para incluir todos os outros analitos e diversas matrizes representativas do grupo de matriz, no mínimo a cada 12 meses, mas preferencialmente a cada 6 meses. |
Os níveis de concentração para determinação das recuperações dos analitos deverão estar na faixa de calibração, ou dentro de um nível de relevância para as amostras analisadas (i.e., os níveis de concentração utilizados na validação do método para a estimativa da Veracidade e Precisão). Esse nível de fortificação deve ser alternado ao longo do tempo. Um dos níveis de concentração deve ser obrigatoriamente o LQ do método.
Os dados obtidos no monitoramento dos valores de Veracidade podem ser utilizados na Verificação concorrente do desempenho do método, conforme o Item VI.2.8.
¶ VI.2.7.1.1 Critérios de Aceitação do Desempenho de Recuperação na Rotina Analítica
Limites aceitáveis para a recuperação individual devem normalmente estar na faixa da recuperação média ± 2 x DPRR obtidos da validação inicial do método ou dos dados de controles de qualidade da rotina (verificação contínua de desempenho do método).
Uma faixa de recuperação (veracidade) de 60 a 140% pode ser aceita para valores de recuperação individuais dos controles de qualidade.
Analitos que possuem histórico de baixas recuperações podem obter valores entre 30 e 60% desde que embasados em dados precedentes, tais como dados de validação e controle de qualidade na rotina (verificação concorrente).
Uma alternativa para esses casos de baixa recuperação é corrigir o valor obtido na amostra pela recuperação do analito, esse valor de recuperação pode ser oriundo dos dados de validação (ou verificação concorrente), ou oriundo da amostra de controle de qualidade do dia no nível de concentração mais próximo da amostra.
Os dados históricos dos valores de veracidade devem demonstrar boa precisão, confirmando o desempenho obtido na validação/verificação inicial do método, assim, analitos com valores de veracidade média fora da faixa de 70 a 120% e DPR acima de 25% devem ser avaliados quanto à manutenção do seu desempenho na rotina analítica.
Caso o resultado dos controles de qualidade obtenha resultado insatisfatórios para um ou mais analito o laboratório deve:
- Reanalisar o lote de amostras e verificar novamente os resultados, ou;
- Não analisar o analito que não obteve resultado satisfatório, neste caso o Relatório de Ensaio deve ser corrigido para adequar ao escopo analisado;
Caso o valor de recuperação dos controles esteja em valores elevados (acima de 140%) e nenhuma amostra testou positivo para este analito com alta recuperação, os resultados podem ser emitidos sem necessidade de reanálise do lote.
Caso o resultado insatisfatório esteja sendo recorrente para uma combinação de analito/matriz, o laboratório deve retirar esse analito do escopo do ensaio.
¶ VI.2.7.1.2 Critérios de Aceitabilidade do Desempenho da Curva de Calibração na Rotina Analítica
Avaliar o coeficiente de correlação (r) estimado, o valor de r deve ser ≥ 0,98;
Recomenda-se avaliar o Desvio Relativo de cada nível de concentração, que este deve ser inferior à ±20%, ou seja, os valores de Recuperação da concentração retrocalculada (Rcr (%)) devem situar entre 80 e 120%.
O analista pode optar por excluir pontos da curva que estejam visualmente deslocados (sugere-se que pontos com desvio maior que 15%), ou indicados como anômalos em testes estatísticos, se atentando para não remover mais do que 22,4% dos pontos.
¶ VI.2.8 Verificação concorrente do desempenho do método (on going)
Os dados gerados no procedimento de controle de qualidade na rotina analítica, conforme descrito no Item VI.2.7 pode ser utilizada para a realização da chamada verificação concorrente da performance do método (on going method performance verification).
As amostras de controle de qualidade do lote devem ser fortificadas em pelo menos na concentração correspondente ao LQ e uma outra em uma concentração superior (2 – 10 x LQ), do mesmo modo que é recomendado para a validação do método.
Os dados históricos de resultado dos controles de qualidade do lote são então utilizados para calcular a veracidade (recuperação) média e o Desvio Padrão Relativo de Reprodutibilidade parcial (DPRR).
A verificação concorrente deve abordar diversos tipos de matriz dentro de um mesmo grupo. A escolha da matriz utilizada no lote de análise deve ser representativa da composição das amostras analisadas, garantindo que, periodicamente, a maioria das matrizes das amostras seja avaliada.
Para utilizar os dados de verificação concorrente para validar o método devem estar presentes os dados de no mínimo 5 lotes de análise, preferencialmente que tenham sido realizadas durante um período não tão curto entre os lotes, por grupo de matriz.
¶ VI.2.9 Sequência de injeção na rotina analítica
A sequência de injeção das amostras, na rotina analítica e nos ensaios de validação do método devem ser construídas de forma a minimizar variações de sinal instrumentais ao longo da injeção do lote de amostras. Por isso toda sequência de injeção deve se iniciar com a curva de calibração, preferencialmente em ordem crescente de concentração, e concluir com a injeção de pelo menos um ponto da curva de calibração, este ponto deve divergir no máximo em 30% do sinal do padrão injetado no início da sequência.
Para sequências com elevado número de amostras, recomenda-se iniciar e finalizar a sequência de injeção com uma curva de calibração e que a curva de calibração seja reinjetada a cada quantidade máxima de amostras (i.e., a cada 20 amostras uma curva de calibração), tal procedimento é chamado de calibração por blocos (braketing calibration). Sendo que a curva de calibração utilizada para quantificar o lote de análise deve ser estabelecida com todos os pontos injetados (i.e., utilizando todas as injeções da curva de calibração injetada no início e no final da sequência de injeção).
¶ VI.2.10 Confirmação de resultado violado
Sempre que análise de uma amostra quantifique alguma suspeita de violação (i.e., a presença de algum agrotóxico acima do LMR, ou agrotóxicos de uso não autorizado (agrotóxico proibido, ou não permitido para a cultura em questão, ou de produção orgânica), levando em conta a incerteza do método, o resultado deve ser confirmado através da reanálise.
O lote de reanalise para confirmação de resultado deve ser preparado conforme a indicação no Item VI.2.7.1., além disso, por se ratar de uma análise de confirmação de violação alguns procedimentos à mais são recomendados:
- Reanalisar a amostra em uma curva de calibração na mesma matriz da amostra reanalisada (nos casos de CCM e CCF);
- Caso a análise inicial tenha sido realizada em uma matriz muito diferente (1) da matriz da amostra que será reanalisada (exemplo: amostra de banana em que a 1a análise foi quantificada em uma CCM de tomate), recomenda-se que, além da ação 1 acima, a reanálise seja realizada em duplicata (duas frações da mesma amostra), visando assim reduzir o impacto do efeito matriz.
- Diferença do Efeito Matriz muito grande entre as duas matrizes, e.g.: diferença maior que 25%.
O resultado após as reanalises deve ser calculado como média dos resultados considerados, sendo duas possibilidades:
- A média entre resultado inicial e resultado da reanalise;
- A média do resultado das reanálises em duplicata (caso b. acima).
Avaliar a precisão dos resultados, sendo que os resultados individuais devem apresentar desvio máximo de 30% em relação à média.
Caso algum resultado tenha variado mais que 30% em relação à média, o laboratório deve analisar a amostra uma 3ª vez e emitir o resultado considerando a média dos dois resultados mais próximos entre si.
Conforme estipulado no Item II.7, é crucial confirmar qualquer resultado violado por meio da avaliação da variação da razão de íons. Recomenda-se que essa variação não ultrapasse 30% em relação ao íon mais intenso. Caso a variação da razão de íons exceda esse limite, o resultado não deve ser emitido, pois as chances de um falso positivo aumentam significativamente. Apesar dessa recomendação geral, a diferença máxima de 30% na razão de íons não deve ser tomada como um limite absoluto e a interpretação automatizada dos dados com base nos critérios, sem interpretação complementar por um analista experiente não é recomendada [19].
Além de avaliar a variação da razão de íons o analista deve verificar se a transição de confirmação apresenta o mesmo perfil cromatográfico do pico da transição de quantificação e o mesmo tempo de retenção ± 0,1 minutos, ou seja, os picos devem se sobrepor completamente.
¶ VI.3 Contaminantes Inorgânicos;
¶ VI.3.1 Validação (Requisitos Mínimos e Critérios de Aceitação)
Os parâmetros a serem calculados durante o processo de validação de métodos para a determinação de contaminantes inorgânicos em atendimento às demandas do PNCRC são:
- Linearidade;
- Seletividade e Efeito Matriz;
- Limite de detecção;
- Limite de quantificação;
- Precisão (repetitividade e reprodutibilidade intralaboratorial);
- Recuperação/veracidade;
- Robustez;
- Incerteza de Medição.
Antes do processamento dos dados obtidos durante a validação, recomenda-se avaliar os desvios padrão relativos entre as leituras de cada replicata e a existência de valores dispersos (outliers).
¶ VI.3.1.1 Linearidade
A faixa de trabalho e a curva de calibração devem ser estabelecidas a partir das concentrações nas quais a linearidade é constatada.
Os padrões utilizados na construção da curva de calibração devem ser preparados numa solução que corresponda tanto quanto possível à da solução da matriz a analisar (por exemplo, relativo à concentração de ácido ou à composição).
¶ VI.3.1.1.1 Procedimento de Estimação da Linearidade
Observar os procedimentos descritos no Item II.2 deste Manual.
Na construção da curva de calibração devem-se usar seis replicatas de cada nível e, pelo menos, cinco níveis de concentração. Cada uma das replicatas deve ser lida três vezes.
Sempre que possível, empregar os níveis equivalentes ao LMC na construção da curva de calibração.
A concentração correspondente ao limite de quantificação deve estar contemplada na curva de calibração. O limite superior da curva é geralmente restringido pela resposta do equipamento, quando possível, o nível de 2,0 LMC deverá ser incluído.
As soluções padrão devem ser preparadas e a curva de calibração deve ser construída da mesma forma como serão empregadas na rotina.
Verificar a ausência de valores discrepantes para cada nível de concentração utilizando testes estatísticos apropriados.
Verificar a homoscedasticidade, a independência e a normalidade da distribuição dos resíduos, conforme recomenda no Item II.2 do manual.
Alternativamente, quando possível, a avaliação da linearidade pode ser realizada a partir dos dados obtidos de, no mínimo, 4 curvas de calibração construídas em dias diferentes. Dessa forma, a avaliação refletirá de maneira mais fidedigna a rotina analítica. Nesse caso, o número de replicatas e leituras poderão ser os mesmos utilizados na rotina.
¶ VI.3.1.1.2 Critérios de Aceitação da Linearidade
Após a aplicação dos testes de valores anômalos e exclusão dos dados indicados avaliar o comportamento da curva de calibração conforme testes de adequações tais como:
- Teste de homoscedasticidade dos resíduos (ex: teste Brown-Forsythe (ou teste de Levene modificado), ou o teste F (Fischer-Snedecor));
- Teste de Independência dos Resíduos (ex: Durbin-Watson);
- Teste de análise de variância (ANOVA) pode ser utilizada para a avaliação da significância da regressão e a significância do erro da regressão.
Descartados os valores discrepantes e, em se tratando da variância homogênea dos resíduos e distribuição normal dos dados (resíduos aleatórios), definir a equação da regressão linear e o coeficiente de determinação, R2, a partir do método dos mínimos quadrados ordinários. Caso os resíduos apresentem distribuição heterocedástica o ajuste da curva analítica deve ser realizado através do método dos mínimos quadrados ponderados, conforme o Item II.2.
O laboratório deve avaliar o grau de dispersão dos resíduos da regressão. Essa avaliação pode ser feita através das ferramentas:
- ANOVA com o teste F para falta de ajuste.
- Recuperação da concentração retrocalculada (Rcr (%)) por ponto que deve ficar entre 85 e 115%.
- Recuperação da concentração retrocalculada (Rcr (%))média por nível de calibração entre 85 e 115% média.
- Análise gráfica visual dos resíduos.
¶ VI.3.1.2 Seletividade e Efeito Matriz
Observar os procedimentos descritos no Itens II.6 Seletividade e Item II.3 Efeito Matriz.
Os experimentos para avaliação da seletividade devem ser conduzidos, no mínimo, para os níveis: 0,2 e 1,0 LMC. Recomenda-se o uso do analito em solvente puro e do analito em matriz branca, dessa forma, parte desse experimento poderá ser utilizada para a extensão de escopo.
¶ VI.3.1.2.1 Critérios de Aceitação da Seletividade e Efeito Matriz
A variação máxima permitida entre a média do valor obtido para cada nível das amostras fortificadas e as soluções padrão não deve ser superior a 10%.
Caso os critérios de aceitação não sejam alcançados, a linearidade, a veracidade e a precisão estarão seriamente comprometidas, sendo neste caso necessário que a curva de calibração seja construída a partir de amostras brancas fortificadas após a digestão.
¶ VI.3.1.3 Limite de Detecção
Limite de detecção é o teor mínimo a partir do qual é possível deduzir a presença do analito com certeza analítica razoável (95% de confiança).
¶ VI.3.1.3.1 Procedimento de Avaliação do Limite de Detecção
Realizar a análise de, no mínimo, 21 replicatas da matriz branca e utilizar os valores obtidos para calcular:
LD = 3,3 s / b
Onde:
s = desvio padrão da resposta instrumental do branco
b = inclinação (coeficiente angular) da curva analítica
Nesse caso, matriz branca é aquela que apresenta concentração que não interfere nos resultados das determinações podendo considerar, mas não se limitar a:
- Concentração de um composto na matriz que resulte num sinal do equipamento muito abaixo ao sinal no LQ;
- Ausência do sinal.
Quando o branco não gera sinal, pode-se adotar para o valor de “s” o desvio padrão (n ≥ 6) da resposta instrumental do menor nível da curva analítica.
¶ VI.3.1.3.2 Critérios de Aceitação do Limite de Detecção
O LD deve ser menor que três décimos do LQ.
¶ VI.3.1.4 Limite de Quantificação
Teor mínimo medido a partir do qual é possível medir o analito com certeza estatística razoável (95% de confiança).
Se a veracidade e a precisão são constantes numa gama de concentrações centrada no limite de detecção, o limite de quantificação é numericamente igual a dez vezes o desvio-padrão da média de ensaios em branco (n > 20).
¶ VI.3.1.4.1 Procedimento para avaliação do Limite de Quantificação
Utilizar os resultados obtidos no ensaio de LD e calcular:
LQ = 10 s / b
s = desvio padrão da resposta instrumental do branco (n > 20)
b = inclinação (coeficiente angular) da curva analítica
Quando o branco não gera sinal, pode-se adotar para o valor de “s” o desvio padrão (n ≥ 6) da resposta do menor nível da curva analítica.
¶ VI.3.1.4.2 Critérios de Aceitação do Limite de Quantificação
Os critérios de aceitação para o LQ são descritos na Tabela 9 abaixo.
Tabela 9: Critério de aceitação do LQ [30].
|
Elemento(s) |
Critério de aceitação |
||
|
Estanho na forma inorgânica |
≤ 10 mg/kg |
||
|
Chumbo |
LMC ≤ 0,02 mg/kg |
0,02 mg/kg < LMC < 0,1 mg/kg |
LMC ≥ 0,1 mg/kg |
|
≤ LMC |
≤ ⅔ do LMC |
≤ ⅕ do LMC |
|
|
Cádmio e mercúrio |
≤ ⅖ do LMC |
≤ ⅖ do LMC |
≤ ⅕ do LMC |
|
Arsênio na forma inorgânica e arsênio total |
LMC ≤ 0,03 mg/kg |
0,03 mg/kg < LMC < 0,1 mg/kg |
LMC ≥ 0,1 mg/kg |
| ≤ LMC |
≤ ⅔ do LMC |
≤ ⅔ do LMC |
|
|
Níquel
|
LMC ≤ 0,3 mg/kg |
0,3 mg/kg < LMC < 0,6 mg/kg |
LMC ≥ 0,6 mg/kg |
|
≤ LMC |
≤ ⅔ do LMC |
≤ 1/3 LMC |
|
A confirmação do valor do LQ deve ser realizada, no mínimo com 6 replicatas, a partir da matriz branca fortificada no respectivo nível do LQ. Os resultados devem alcançar valores de recuperação de acordo com a Tabela 10 [33]:
Tabela 10: Limites para valores aceitáveis de recuperação.
|
Fração mássica |
Intervalo |
|
≤ 1 μg/kg |
-50% a +20% |
|
> 1 μg/kg a 10 μg/kg |
-30% a +20% |
|
≥ 10 μg/kg |
-20% a +20% |
¶ VI.3.1.5 Precisão
A precisão do método é avaliada por meio da repetitividade e da reprodutibilidade interna.
¶ VI.3.1.5.1 Procedimentos para Avaliação da Repetitividade
Preparar, no mínimo, seis replicatas de matrizes brancas fortificadas em dois níveis de concentração: 0,2 – 1,0 TMC. Realizar o experimento, no mínimo, por duas vezes em condições de repetitividade.
Calcular, a partir dos resultados obtidos, a concentração de cada amostra, a concentração média para cada nível de adição, o desvio-padrão (sr) e o desvio-padrão relativo em condições de repetitividade (RSDr) que deve determinado conforme as recomendações descritas no Item II.4.4.
Calcular o desvio-padrão relativo teórico para cada nível de concentração utilizando a equação de Horwitz:
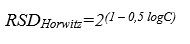
Onde:
C é a fração mássica expressa sob a forma de uma potência de dez, i.e., Concentração 1 µg kg-1 corresponde à 10-9.
Calcular o valor de HORRAT em condições de repetitividade, HORRATr, segundo a equação:
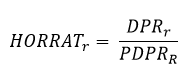
Onde:
PDPRR : (Desvio-padrão relativo predito) = 2 C-0,15
C = Concentração (µg kg-1) × 10-9
Calcular o limite de repetitividade, r, segundo a equação:
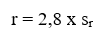
¶ VI.3.1.5.1.1 Critérios de Aceitação da Repetitividade
No caso de análises de amostras em condições de repetitividade, o desvio-padrão relativo (DPRr) da média deve ser menor que dois terços do valor do DPRHorwitz.
Os valores HORRATr devem ser inferiores a 2.
¶ VI.3.1.5.2 Procedimentos para Avaliação da Reprodutibilidade Interna
Preparar, no mínimo, seis replicatas de amostras em dois níveis de concentração: 0,2 – 1,0 LMC.
Repetir o experimento por, pelo menos, duas vezes em dias diferentes em condições de reprodutibilidade.
Calcular a concentração obtida para cada replicata e a concentração média, o desvio-padrão e o desvio-padrão relativo em condições de reprodutibilidade interna (DPRR), para cada nível, que deve determinado conforme as recomendações descritas no Item II.4.4.
Calcular o valor de HORRAT em condições de reprodutibilidade intralaboratorial, HORRATR, segundo a equação:
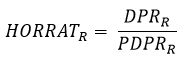
Onde:
PRSDR (Desvio-padrão relativo predito) = 2 C-0, 15
C = Concentração (µg kg-1) × 10-9
Calcular o limite de reprodutibilidade, R, segundo a equação:
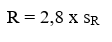
¶ VI.3.1.5.2.1 Critérios de Aceitação para Reprodutibilidade Intralaboratorial
No caso de análises de matrizes brancas fortificadas em condições de reprodutibilidade intralaboratorial, o desvio-padrão relativo (DPR) intralaboratorial dos resultados não deve exceder o valor de DPRHorwitz.
Os valores HORRATR devem ser inferiores a 2.
¶ VI.3.1.6 Procedimentos para Estimação da Veracidade/Recuperação utilizando Material de Referência (MR) ou o Material de Referência Certificado (MRC)
O MRC ideal deve apresentar concentrações similares ao TMC na matriz de interesse.
Quando um MRC apropriado não está disponível, podem ser utilizados materiais de referência (MR) obtidos a partir de ensaios de proficiência. No caso do emprego do MR, utilizar a incerteza do valor estimado por consenso.
Analisar, no mínimo, seis replicatas das amostras de MR/MRC em conformidade com as instruções para o método.
Calcular a média, o desvio-padrão e o desvio-padrão relativo.
Calcular a veracidade pela equação:
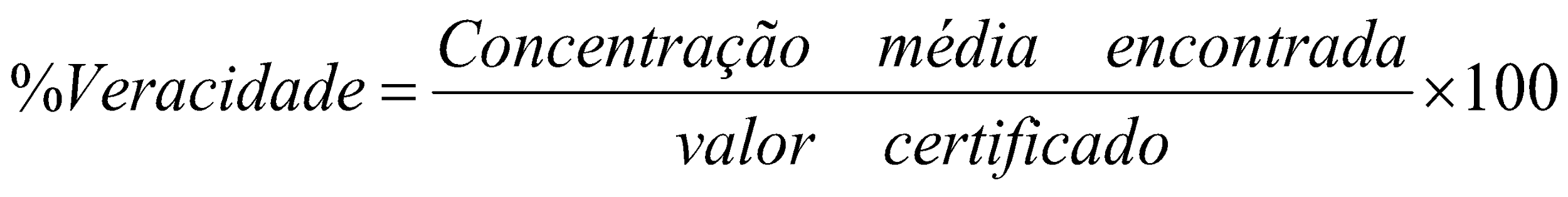
A veracidade é verificada comparando-se a média dos resultados obtidos para o MR/MRC, com o valor aceito como verdadeiro, μ, conforme procedimento a seguir:
Calcular a incerteza do processo de medição (expressa pelo seu desvio-padrão), σD, pela equação:
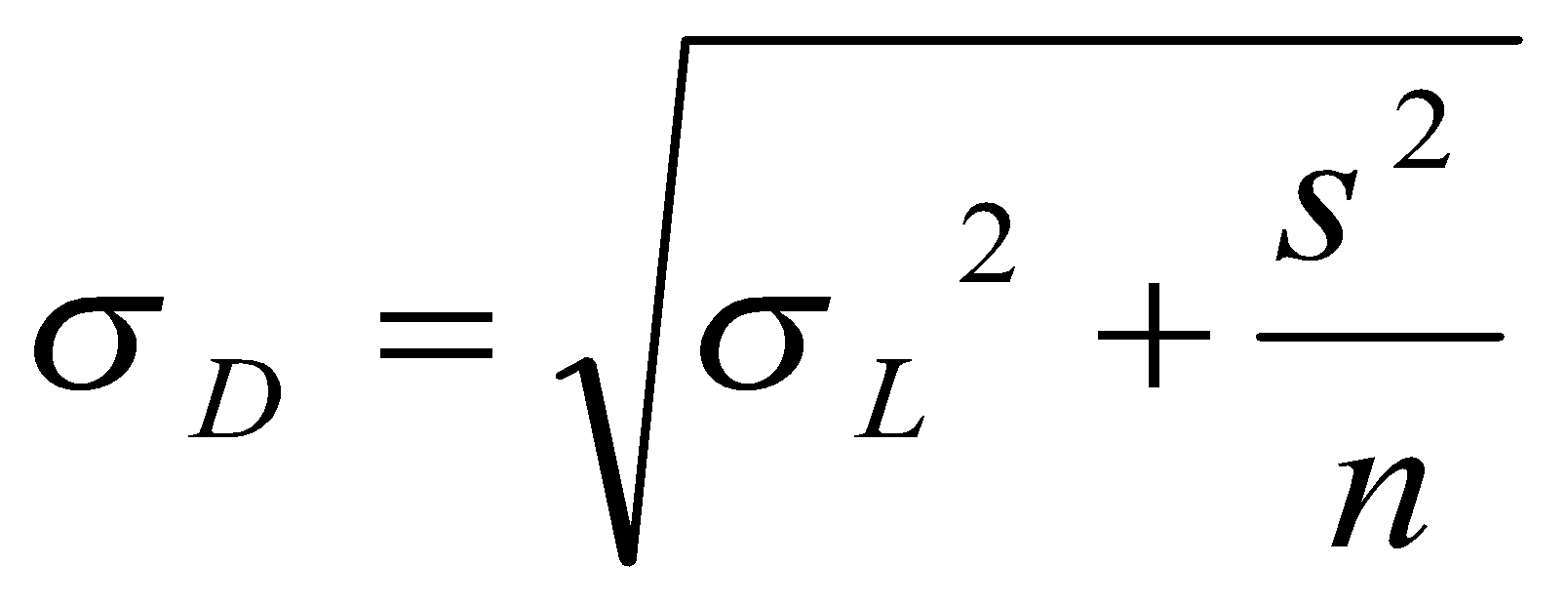
Onde:
σL é a incerteza do MR/MRC informada e S é o desvio-padrão das medidas.
¶ VI.3.1.6.1 Critérios de Aceitação da Veracidade Empregando MRC/MR
Se:
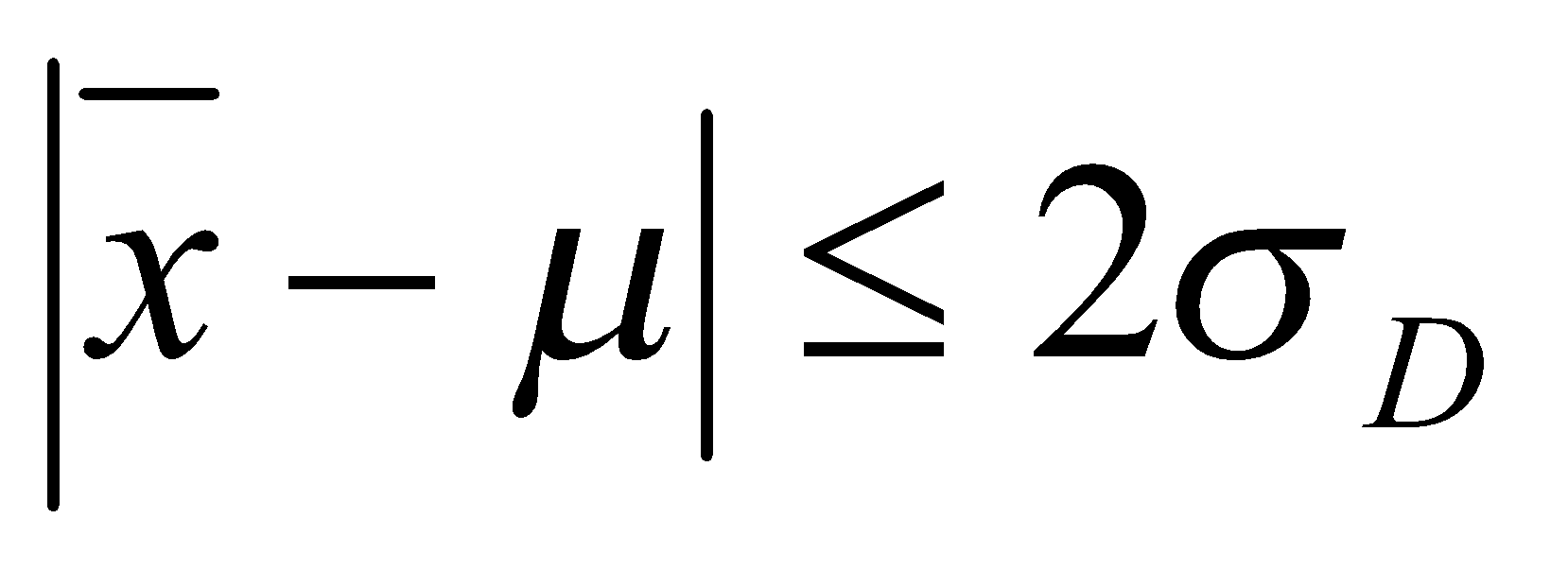
o processo de medição apresenta a veracidade adequada.
Se:
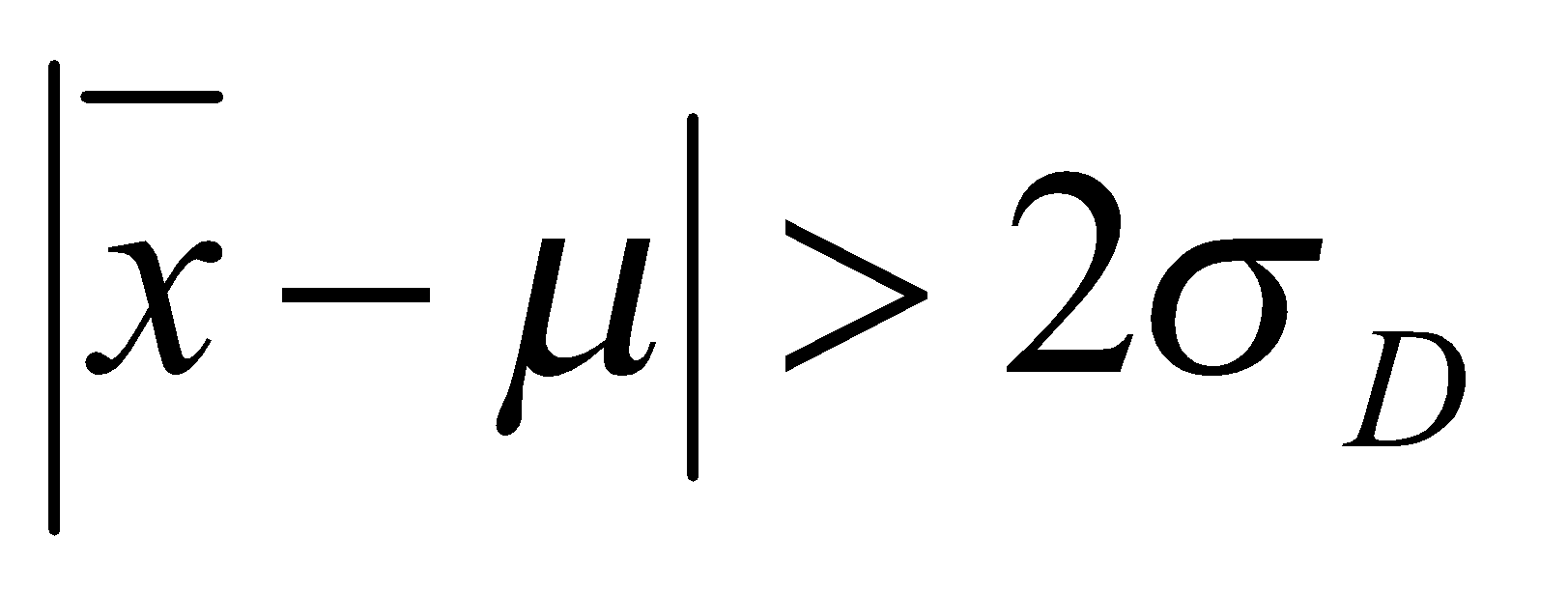
o processo de medição não apresenta a veracidade adequada.
¶ VI.3.1.7 Procedimentos para Estimação da Precisão (Repetitividade) utilizando Material de Referência (MR) ou o Material de Referência Certificado (MRC)
A repetitividade de um processo de medição obtida a partir de um MR ou MRC é avaliada comparando-se o desvio-padrão das medidas (s), com o valor requerido do desvio-padrão intralaboratório (⌠wo).
O σwo pode ser calculado a partir da fórmula:
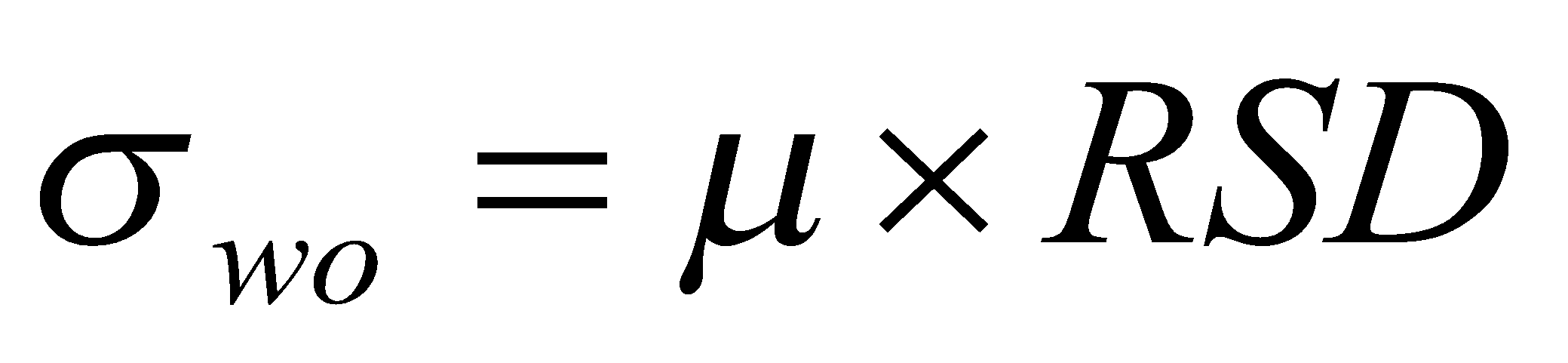
Onde:
µ é o valor de concentração estabelecido para o MR / MRC e RSD é o desvio-padrão relativo aceitável para a faixa de fração mássica de acordo com a Equação de Horwitz descrita no Item VI.3.1.5.
Calcular a seguinte razão:
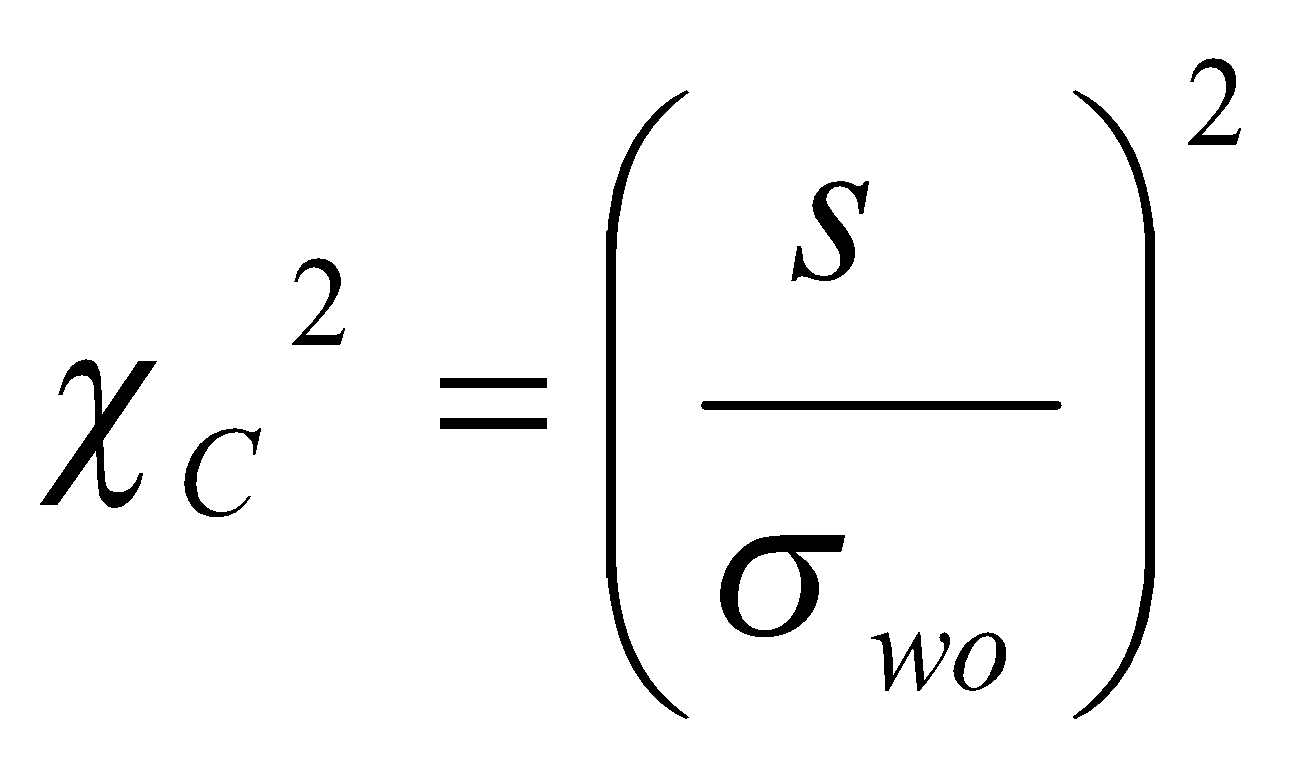
E comparar com o respectivo valor tabelado, de acordo com a equação:
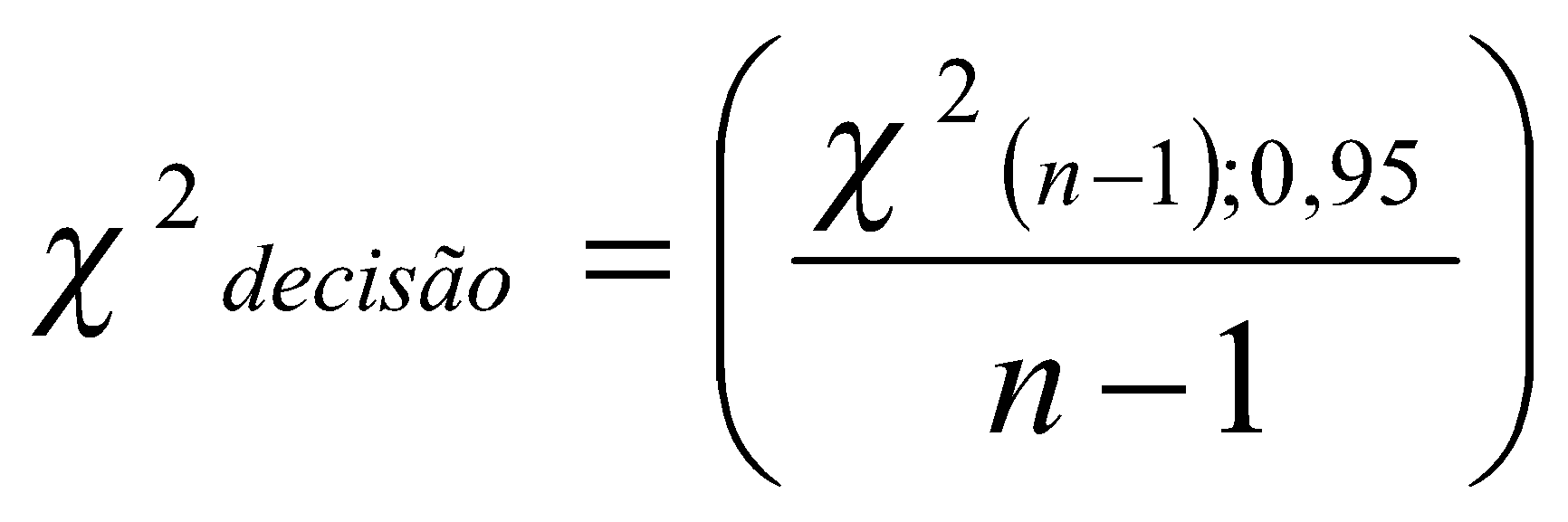
Na tabela usar o valor da área de probabilidade de 1 - 0,95, ou seja, 0,05 (ou 0,95 ) quantil da distribuição (x2).
¶ VI.3.1.7.1 Critérios de Aceitação da Precisão (Repetitividade) Calculada com o Uso de MR e MRC.
Se xc2 ≤ x2decisão, o processo de medição apresenta a precisão intralaboratorial requerida.
Se xc2 > x2decisão, o processo de medição não apresenta a precisão intralaboratorial requerida.
Quando os MRCs não estiverem disponíveis, se aceita que a veracidade das medições seja avaliada por meio da recuperação de quantidades conhecidas do analito adicionadas às matrizes brancas utilizando os dados obtidos no estudo de precisão com amostras fortificadas descrito no Item VI.3.1.5.
Chama-se a atenção para o fato de que quando, ao contrário da contaminação natural, o analito adicionado não se encontra ligado quimicamente à matriz real, os resultados obtidos com esse método são menos fiáveis.
¶ VI.3.1.8 Procedimentos para a Avaliação da Recuperação/Veracidade
Utilizar os dados obtidos no ensaio de reprodutibilidade interna.
Calcular a recuperação média por cada nível de concentração.
¶ VI.3.1.8.1 Critérios de Aceitação da Recuperação
A faixa aceitável para os valores de recuperação está escrita na Tabela 10.
¶ VI.3.1.9 Procedimentos para a Avaliação da Robustez
Para avaliação da robustez do método recomenda-se o uso de ferramentas de planejamento fatorial.
No Anexo IV está presente um exemplo do uso do planejamento fatorial fracionário conhecido como Teste de Youden, para verificar a robustez de um método de quantificação de Contaminantes Inorgânicos em alimentos.
¶ VI.3.1.9.1 Critérios de Aceitação da Robustez
Conforme o Teste de Youden descrito no Anexo IV, avaliar os resultados obtidos.
O método será robusto se Ftabelado for maior que Fcalculado;
O SDi deve ser menor que o desvio-padrão obtido em condições de reprodutibilidade intralaboratorial.
A avaliação entre o SDi e o desvio-padrão do método em condições de reprodutibilidade pode ser realizada pelo teste F [5].
Sempre que o SDi for significativamente maior que o desvio-padrão do método, em condições de reprodutibilidade intralaboratorial, pode deduzir-se que o conjunto dos fatores têm influência no resultado, mesmo que cada fator isoladamente não influencie significativamente, e que o método não é suficientemente robusto face às alterações ensaiadas;
Se o valor de algum efeito ultrapassar a linha de significância do gráfico de barras, pode-se considerar que o mesmo é significativo e recomenda-se implantar um controle nesta etapa do processo. Caso seja necessário, recomenda-se aprofundar o estudo sobre a influência dos fatores significativos realizando–se um planejamento fatorial completo para eles.
¶ VI.3.2 Procedimentos para a Avaliação da Incerteza de Medição
Recomenda-se estimar a incerteza de medição segundo procedimento estabelecido no Item II.12.
¶ VI.3.2.1 Critérios de Aceitação da Incerteza de Medição
Os procedimentos analíticos aplicados ao controle oficial devem produzir resultados cujas incertezas de medição padrão sejam inferiores à incerteza de medição padrão máxima calculada por meio da seguinte fórmula [30]:
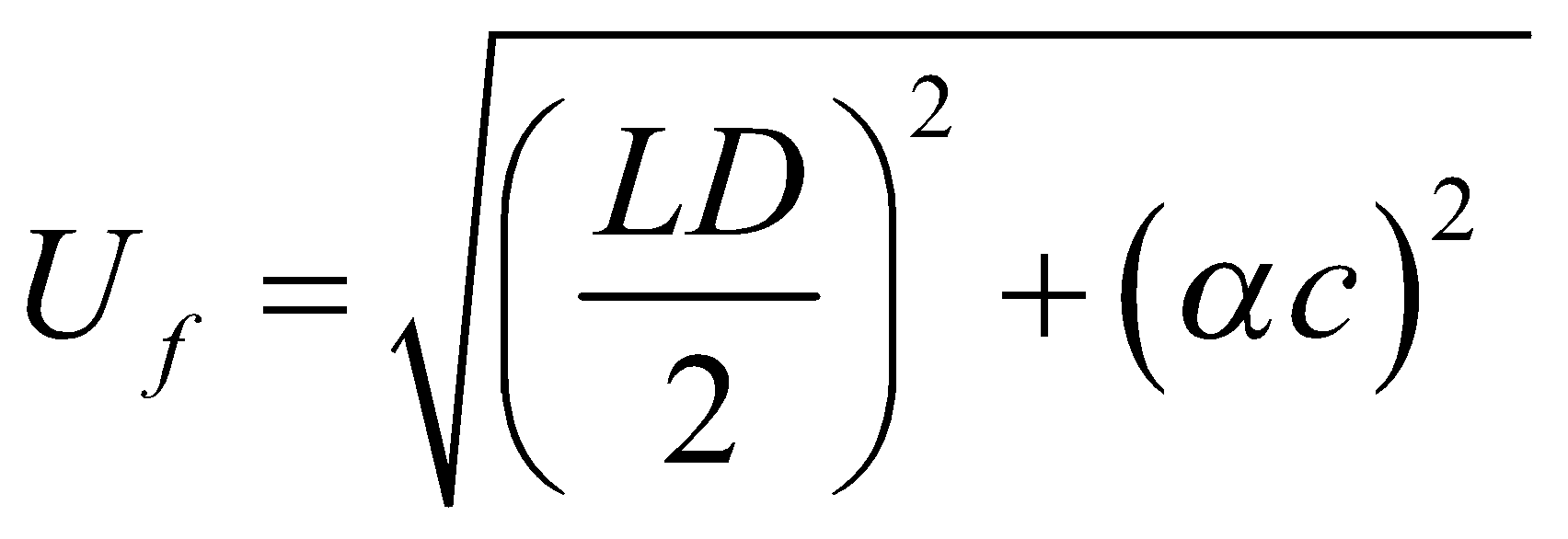
Onde:
Uf = incerteza de medição padrão máxima (µg kg-1).
LD = limite de detecção do método (µg kg-1).
c = corresponde à concentração em causa (µg kg-1).
α = é um fator numérico cuja utilização depende de c.
Os valores de α a serem utilizados se encontram na Tabela 11 abaixo:
Tabela 11: Valores de α a serem utilizados de acordo com a concentração.
|
c (µg kg-1) |
α |
|
≤ 50 |
0,2 |
|
51 a 500 |
0,18 |
|
501 a 1000 |
0,15 |
|
1001 a 10 000 |
0,12 |
|
> 10 000 |
0,1 |
¶ VI.3.3 Procedimentos para Extensão de Escopo (Novas Matrizes)
Para a inclusão de novas matrizes em procedimentos analíticos validados devem ser avaliados, no mínimo, os parâmetros: Efeito Matriz, Veracidade/Recuperação e Repetitividade. Os estudos devem ser conduzidos nos níveis de 0,2 e 1,0 TMC referentes à matriz nova.
Para avaliação do efeito de matriz realizar o procedimento descrito no item VI.3.1.2. Para avaliação da Veracidade/Recuperação realizar o procedimento descrito no item VI.3.1.8 e para a Repetitividade realizar o procedimento descrito no item VI.3.1.5.1.
Observação: Em alguns casos, mesmo os experimentos sendo conduzidos em sua totalidade como proposto acima, recomenda-se a avaliação da recuperação / veracidade com o uso de MRC/MR referente à matriz nova. Tal recomendação visa garantir que a aplicação do procedimento analítico não é influenciada pelas espécies químicas (orgânicas e inorgânicas) presentes em amostras naturalmente contaminadas, ou mesmo possíveis interações entre o analito e a matriz. Um exemplo de procedimento analítico que apresenta forte influência das espécies químicas é a determinação de arsênio total em pescados.
¶ VI.3.3.1 Critérios de Aceitação da Extensão de Escopo (Novas Matrizes)
Utilizar os mesmos critérios de aceitabilidade estabelecidos para a validação completa do método, conforme descrito nos itens VI.3.1 ao VI.3.8.
¶ VI.4 Micotoxinas
Devido à ocorrência natural de diferentes micotoxinas em um mesmo produto de origem vegetal ou animal, os procedimentos analíticos multitoxinas são preferencialmente recomendados.
Quando o procedimento analítico for destinado à aplicação a mais de uma matriz, de uma mesma classe ou de classes distintas de produtos, deve ser realizada uma validação completa para a matriz representativa. Em adição, validações curtas devem ser conduzidas objetivando avaliar o comportamento do procedimento analítico para as demais matrizes do grupo, de forma a garantir que o procedimento analítico seja aplicável de maneira integral a todas as matrizes que fazem parte do escopo conforme procedimentos descritos neste item.
De maneira sistemática, os parâmetros que impactam na qualidade analítica precisam ser monitorados, conforme descrito na Parte IV, de modo a garantir durante a rotina analítica que o procedimento analítico continua operando dentro dos parâmetros preconizados.
¶ VI. 4.1 Matrizes Representativas
Para utilização de matriz representativa, os seguintes critérios devem ser observados:
- No mínimo uma matriz representativa por grupo de produtos deve ser avaliada durante a validação.
- Quando um procedimento analítico for implementado na rotina para uma variedade de matrizes de um mesmo grupo, dados complementares deverão ser coletados de forma a consolidar a validação concorrente para todas as matrizes daquele grupo.
- Na seleção da matriz representativa, devem ser utilizados os critérios estabelecidos na Tabela 12;
- Matrizes representativas podem ser utilizadas na validação de procedimentos analíticos mono e multitoxinas.
- A Tabela 12 apresenta as classes de matrizes representativas para cada grupo de micotoxinas de interesse, que podem ser avaliadas como uma combinação de matriz x micotoxina durante a validação de procedimentos analíticos.
- Todas as micotoxinas contempladas pelo escopo do procedimento analítico necessitam ser validadas.
- Preferencialmente, cada batelada de amostras a serem analisadas deve ter uma curva de calibração contemplando todas as micotoxinas do escopo do procedimento analítico.
- Todas as micotoxinas devem ser contempladas na curva de calibração.
- Sempre que uma substância ultrapassar o nível máximo requerido (não conformidade) a amostra deve ser reanalisada, de forma a atender aos requisitos estabelecidos na Parte IV deste Manual.
Tabela 12: Grupo de matrizes, por categoria, a serem utilizadas na validação de procedimentos analíticos e no controle de qualidade para micotoxinas.
|
Grupo |
Propriedades comuns |
Categoria |
Matrizes pertencentes à mesma Categoria. |
|
I |
Elevado teor de óleo |
Frutos de casca rija Sementes oleaginosas e seus subprodutos Frutos oleaginosos e seus produtos |
Amendoim, avelãs, castanha-do-brasil, pistache, nozes, amêndoas em geral e suas respectivas pastas ou manteigas e coco Sementes de girassol, sementes de algodão |
|
II |
Elevado teor de amido e/ou de proteínas e baixo teor de água e de gordura |
Cereais e seus produtos dietéticos Leguminosas |
Arroz, milho, cevada centeio, trigo, farinha de milho, trigo, farinha de trigo Feijão, lentilhas |
|
III |
Alimentos para animais | Ração e Ingredientes de ração |
Farelo de soja, farelo de trigo, farelo de milho, ração de frango, de suíno, equino, bovina |
|
IV |
Elevado teor de açúcar e baixo teor de água | Frutas secas |
Uva passa, ameixa, damasco, tâmara, figo, banana, dentre outras |
|
V |
Produtos únicos | Café verde |
Café verde |
| Café torrado e solúvel |
Café torrado ou solúvel |
||
|
VI |
Leite e produtos lácteos | Leite, queijo, produtos lácteos |
Leite fluído, leite em pó, iogurte, natas |
|
VII |
Elevado teor de água |
Sucos de frutas Bebidas alcoólicas |
Suco de uva, suco de maçã, sidra, cerveja e vinho |
|
VIII |
Proteína de origem animal | Miúdos, músculos, produtos cárneos |
Fígado, rim, presunto |
¶ VI.4.2 Validação (Requisitos Mínimos e Critérios de Aceitação)
A validação de procedimentos analíticos para determinação de micotoxinas em matrizes de origem vegetal ou animal, deverá ser conduzida atendendo aos critérios mínimos dos seguintes parâmetros de desempenho:
- Seletividade;
- Efeito Matriz;
- Linearidade (Curva de Calibração);
- Veracidade e Precisão
- Limite de Detecção (LD) e Limite de Quantificação (LQ);
- Robustez;
- Estudo de Estabilidade;
- Incerteza de Medição;
¶ VI. 4.2.1 Seletividade
Para se avaliar a presença de possíveis interferentes, recomenda-se que tal avaliação seja feita em amostras “brancas”, no nível do LQ e, se possível, no nível do TMC para cada matriz representativa do escopo do método. Testes preliminares podem ser feitos livremente com base nos requisitos descritos a seguir, mas a evidência objetiva deve ser realizada em ao menos 6 replicações independentes. Pode-se utilizar os dados obtidos na etapa de avaliação da precisão e veracidade do método.
De acordo com o documento SANTE/12089/2016 [20], a cromatografia combinada com a espectrometria de massas é o método de escolha para a identificação de micotoxinas. Alternativamente, pode-se aplicar cromatografia líquida com detecção por fluorescência, mas apenas quando uma etapa de purificação baseada em imunoafinidade específica para a(s) micotoxina(s) alvo tiver sido empregada.
Os critérios abaixo fornecidos são recomendações que são desejadas para alcançar uma identificação adequada. Durante a validação do método, verificar se os critérios são atendidos dentro da faixa de trabalho do método, usando amostras fortificadas ou materiais de referência certificados. Além disso, deve-se verificar que, para amostras em branco, não ocorram identificações falso-positivas.
¶ VI 4.2.1.1 Critérios de Aceitabilidade
¶ VI 4.2.1.1.1 Requisitos para Cromatografia
Recomenda-se que o tempo de retenção mínimo aceitável para o analito em exame deve ser o dobro do tempo de retenção correspondente ao volume morto da coluna.
O tempo de retenção do analito no extrato da amostra deve corresponder ao da média dos padrões de calibração medidos na mesma sequência, com uma tolerância de ±0,2 minutos ou ±50% da largura do pico à meia altura (o que for maior), tanto para cromatografia gasosa quanto para cromatografia líquida. Com UHPLC e GC, as variações são tipicamente de ±0,1 minutos.
Caso um analito isotopicamente marcado com 13C (padrão interno) tenha sido adicionado à amostra ou ao extrato, o tempo de retenção do analito deve corresponder ao de seu padrão interno marcado com uma tolerância de ±0,05 minutos.
¶ VI 4.2.1.1.2 Requisitos para Detecção por Fluorescência
Isso se aplica a moléculas que exibem fluorescência nativa e a moléculas que exibem fluorescência após transformação ou derivatização. A seleção dos comprimentos de onda de excitação e emissão, em combinação com as condições cromatográficas, deve ser feita de maneira a minimizar a ocorrência de componentes interferentes nos extratos de amostras em branco. O pico máximo mais próximo no cromatograma deve ser separado do pico do analito designado por pelo menos uma largura de pico completa a 10% da altura máxima do pico do analito.
¶ VI 4.2.1.1.3 Requisitos para Detecção por Espectrometria de Massas
A identificação depende da seleção adequada de íons que sejam seletivos para o analito. Íons moleculares ou moléculas (des)protonadas (ou, se não disponíveis, adutos) devem ser incluídos no procedimento de medição e identificação sempre que possível. Fragmentos de baixa massa (m/z < 100) e íons fragmentados que resultam da perda de água ou grupos comuns são frequentemente menos específicos e, portanto, podem ser menos úteis para fins de identificação.
A identificação deve ser baseada em picos cromatográficos observados nos cromatogramas de íons extraídos de dois ou mais íons (ver tabela a seguir) que sejam específicos para o analito. Os picos devem ter forma semelhante, sobreposição entre si, e a razão dos íons (definida como a resposta do pico com menor área dividida pela resposta do pico com maior área) deve estar dentro de ±30% (relativo) da obtida a partir da média dos padrões de calibração da mesma sequência. Quando um cromatograma de íons extraído mostra evidência de interferência significativa, ele não deve ser utilizado para identificação.
Além do grau de seletividade dos íons medidos, diferentes tipos e modos de detecção por espectrometria de massas fornecem diferentes graus de seletividade (ou até especificidade), o que se relaciona com a confiança na identificação. Os requisitos para identificação são fornecidos na Tabela 13 a seguir. Eles devem ser considerados como critérios de orientação, e não como critérios absolutos para provar a presença ou ausência de um analito.
Tabela 13: Requisitos de identificação de micotoxinas para diferentes técnicas de espectrometria de massas (EM).
|
Características do Analisador de Massas |
Sistemas típicos (exemplos) |
Aquisição |
Requisitos para identificação |
|
|
Número mínimo de íons |
Outros |
|||
|
Resolução de massa unitária |
Quadrupolo (Q), íon trap, Tempo de voo (TOF) |
Full Scan, SIM, faixa limitada de m/z |
3 íons |
Sinal/Ruído ≥ 3d Picos dos analitos nos cromatogramas de íons extraído devem se sobrepor completamente. Razão de íons deve estar dentro da faixa ±30% se comparados a média dos padrões de calibração obtidos na mesma sequência de injeção. |
|
Sequencial (MS/MS) |
Triplo quadrupolo (QQQ), ion trap, Q-trap, Q-TOF, Q-Orbitrap |
MRM, SRM, resolução de massa do íon precursor igual ou melhor que resolução unitária |
2 íons produto |
|
|
Medição de massa exata |
EM de alta resolução: (Q-)TOF (Q-)Orbitrap FT-ICR-MS Setor Magnético |
Full Scan, SIM, faixa limitada de m/z, fragmentação com ou sem seleção de íon precursor ou quaisquer combinações disso |
2 íonsa,b com massa exata: ≤ 5 ppm para m/z maiores que 200 ≤ mDa para m/z menores que 200 |
|
|
Combinação entre EM de estágio único e sequencial (MS/MS) com massa do íon precursor igual ou melhor que resolução unitária |
2 íons: 1 íon molecular, molécula (des)protonada ou íon aduto com massa exata ≤ 5 ppm (ou ≤ 1 mDa para m/z ≤ 200) Mais 1 íon produto MS/MSc |
|||
a) Preferencialmente incluir o íon molecular, molécula (des)protonada ou íon aduto.
b) Incluir ao menos um íon fragmento.
c) Não há requisito específico para exatidão de massa.
d) Em casos em que não haja ruído, o sinal do analito deve estar presente em ao menos 5 aquisições subsequentes.
¶ VI 4.2.2 Efeito Matriz
Curvas de calibração em solução só poderão ser utilizadas se for comprovada a ausência de efeito matriz.
Os procedimentos analíticos que utilizam colunas de imunoafinidade na etapa de purificação são considerados, a princípio, sem efeito matriz, tendo em vista a especificidade de separação das micotoxinas em um gel (coluna de fase sólida de imunosorbente) contendo anticorpos específicos para às micotoxinas pesquisadas para as matrizes pertencentes ao mesmo grupo da Tabela 12. Entretanto, é preciso, avaliar a capacidade de ligação da coluna de imunoafinidade empregada (ng de toxinas) e as condições analíticas do método para classes de matrizes e grupos de micotoxinas.
Quando o procedimento analítico em validação estiver destinado à análise de diferentes matrizes para quantificação das mesmas micotoxinas pode-se, na etapa de avaliação do efeito matriz, fazer uma composição das diferentes matrizes do mesmo grupo conforme Tabela 12.
Se o efeito matriz for comprovadamente positivo na matriz composta, deve-se avaliar cada matriz individualmente, caso contrário, a matriz composta pode ser utilizada nos testes de validação e inclusive como amostra branca nos controles de qualidade de resultados analíticos. Valida-se, então, o procedimento analítico para uma matriz composta e insere-se nos trabalhos de rotina, amostras de controle de qualidade para cada matriz do grupo contaminadas (MR, MRC) ou fortificadas no nível requerido. Faz-se necessário a confirmação do LQ para todas as matrizes.
¶ VI 4.2.2.1 Procedimento de Determinação do Efeito Matriz
Preparar uma curva de calibração CCS com no mínimo 5 níveis de concentração, usando soluções padrão de calibração, não matrizadas, de analito puro em solvente puro.
Analisar, usando a CCS, amostras elaboradas no mínimo em 3 níveis de fortificação, de tal forma que o limite permitido se encontre preferencialmente na região central dos níveis. Um mínimo de 5 réplicas por nível de fortificação de:
- Analito em solvente puro (amostra não matrizada);
- Analito em matriz branca ou em extrato/digerido de matriz branca (amostra matrizada).
Após as análises replicadas dessas amostras matrizadas e não matrizadas, realizadas usando a CCS, proceder à avaliação dos resultados das concentrações do analito obtidas nas várias fortificações analisadas como indicado a seguir.
¶ VI 4.2.2.2 Procedimentos de Avaliação e Critérios de Aceitação do Efeito Matriz
Realizar avaliação do efeito matriz conforme descrito no Item II.3 deste manual utilizando a comparação por níveis de concentração.
Verificar a significância da presença do Efeito Matriz, através do Teste t para médias [5].
¶ VI 4.2.3 Linearidade
Os procedimentos relacionados à curva de calibração devem ser realizados conforme descrito no Item II.3.
A ponderação deve ser realizada. Os pesos (wi) podem ser iguais à: 1/x, 1/x2 ou Σisi2/nsi2.
A avaliação da linearidade deve ser feita comparando a concentração retro calculadas com a concentração nominal da curva (conforme a Recuperação da concentração retrocalculada (Rcr (%)) descrita no item II.2.2).
Os laboratórios devem estabelecer internamente critérios de aceitabilidade dos parâmetros da curva de calibração: dispersão dos resíduos individuais da curva e coeficiente de correlação.
¶ VI 4.2.3.1 Critérios de Aceitabilidade da Regressão
A Recuperação da concentração retrocalculada (Rcr (%)) de cada ponto da curva de calibração não deve variar fora da faixa de 85 a 115% (ou Desvio mais que 15%) comparada a concentração nominal [27].
¶ VI 4.2.4 Veracidade e Precisão (Repetitividade e Reprodutibilidade Interna)
¶ VI 4.2.4.1 Procedimento de Determinação da Veracidade
Analisar seis réplicas de material de referência certificado – MRC ou de matriz branca fortificada com os padrões de calibração, em no mínimo 3 níveis de concentração (alta, média e baixa).
Para as substâncias permitidas os limites de interesse estabelecidos (LMR) devem se encontrar preferencialmente na região central dos níveis de concentração da curva de calibração.
Calcular a recuperação média por nível de fortificação.
A veracidade poderá também ser realizada por comparação dos resultados obtidos com outro procedimento analítico de ensaio previamente validado ou a partir dos resultados da participação em ensaios de proficiência.
Os resultados devem ser considerados em termos de recuperação do valor certificado ou atribuído.
¶ VI 4.2.4.2 Procedimento de Determinação da Precisão - Repetitividade
Preparar e analisar um conjunto de amostras constituídas de matrizes brancas fortificadas ou de MRCs, no mínimo em três (3) níveis de concentração, com as substâncias a analisar, de tal forma que os limites permitidos se encontrem preferencialmente na região central dos níveis de concentração e o LMR seja contemplado, conforme se trate de substâncias permitidas ou banidas.
Para cada nível, a análise deve ser realizada em, pelo menos, seis réplicas independentes. Calcular a concentração determinada para cada amostra replicada.
Calcular as concentrações médias, os desvios-padrão de repetitividade (sr) e os coeficientes de variação de repetitividade (%) das amostras fortificadas em cada nível de concentração.
¶ VI 4.2.4.3 Procedimento de Determinação da Precisão - Reprodutibilidade Interna
Repetir o procedimento adotado para o estudo de repetitividade pelo menos mais duas vezes em dias diferentes, variando sempre que possível os operadores, os instrumentos, as condições ambientais, lotes de reagentes e solventes, entre outros fatores experimentais. Assim o estudo da precisão constitui-se essencialmente de repetidos estudos de repetitividade em diferentes condições experimentais dentro de um laboratório.
Calcular a concentração detectada para cada amostra replicada.
Calcular a concentração média, o desvio-padrão e o desvio-padrão relativo global, para cada nível.
¶ VI.4.2.4.4 Estimando a Veracidade e Precisão
Realizar a estimativa da Veracidade e Precisão (Repetitividade e Reprodutibilidade Interna) conforme descrito no Item II.4.
¶ VI 4.2.4.5 Critérios de Aceitabilidade
O procedimento analítico deve apresentar recuperação média entre 70 e 120 %. Excepcionalmente, a recuperação abaixo do requisito estabelecido poderá ser aceitável desde que devidamente justificado, mas deve situar-se entre 50-130% e apenas quando os critérios de precisão para DPRr e DPRR forem atendidos.
O DPRr (desvio padrão relativo de repetibilidade) deve ser ≤ 20%.
O DPRR (desvio padrão relativo de reprodutibilidade interna) deve ser ≤ 25%.
Esses critérios se aplicam a todas as concentrações.
Caso o teor máximo se aplique a uma soma de toxinas, os critérios de precisão aplicam-se tanto à soma como às toxinas individuais.
¶ VI 4.2.5 Limites de Detecção (LD) e Limite de Quantificação (LQ);
Os procedimentos relacionados à determinação de Limites de Detecção (LD) e Limite de Quantificação, (LQ), devem ser realizados em consonância com o disposto nos itens II.5.1 e II.5.2.
Após a estimativa matemática do Limite de Quantificação (LQ), o mesmo deve ser confirmado em termos de precisão, veracidade e incerteza, conforme requisito específico para o nível em que o LQ se encontra, utilizando materiais de referência ou matrizes brancas fortificadas com padrão.
Analisar no mínimo seis amostras da matriz branca fortificada de preferência de origens diferentes. A relação sinal/ruído deve ser superior a seis (S/R ≥ 6).
Avaliar a aceitabilidade do valor de LQ conforma a Tabela 14 abaixo.
Tabela 14: Critérios de Aceitação do Limite de Quantificação
|
Micotoxina |
Gênero Alimentício |
Requisito LQ (µg/kg) |
|
Aflatoxina B1 |
Alimentos para bebés e alimentos transformados à base de cereais destinados a lactentes e crianças pequenas e alimentos para fins medicinais específicos destinados a lactentes e crianças pequenas |
≤ 0,1 |
|
Aflatoxina B1, B2, G1, G2, cada uma das aflatoxinas |
Todos os outros géneros alimentícios |
≤ 1 |
|
Ocratoxina A |
Produtos de confeitaria de alcaçuz contendo < 97 % de extrato de alcaçuz em base seca |
≤ 10,0 |
|
Cacau em pó |
≤ 3,0 |
|
|
Alcaloides de Ergot/Cravagem (cada um dos 12 epímeros incluídos na soma que definem os limites máximos definidos no regulamento UE 915/2023 |
Cerais e alimentos à base de cereais |
≤ 4 |
|
Alimentos processados a base de cereais para bebês e crianças. |
≤ 2 |
Em todos os outros casos o LQ deve ser ≤ 0,5 x LMR, preferencialmente ser ≤ 0,2 x LMR.
Nos casos em que os limites máximos são aplicáveis a soma de toxinas, então o LQ para cada toxina individual deve ser ≤ (0,5 x LMR)/n, com n sendo o número de toxinas incluídas na definição do LMR.
¶ VI 4.2.6 Estudo de Robustez, Escopo e Portabilidade
Os procedimentos relacionados a determinação de Robustez, devem ser realizados em consonância com o disposto no Item II.8.
A portabilidade é a possibilidade de o procedimento analítico ser transportado/transferido para outro local de execução ou outro equipamento sem, no entanto, perder suas características metrológicas e seu desempenho. Os parâmetros a serem avaliados para demonstrar portabilidade são: linearidade, veracidade, precisão, LQ.
¶ VI 4.2.7 Estudos de Estabilidade (Orientação Geral)
Os procedimentos relacionados a estudos de estabilidade devem ser realizados em consonância com o disposto na Parte III.
O estudo de estabilidade deve ser realizado, preferencialmente, em materiais considerados homogêneos. Deve simular as condições nas quais o laboratório submete as amostras e padrões. Por exemplo, se o laboratório armazena as amostras congeladas por 24 horas ele deve provar a estabilidade da micotoxina nas amostras nesse período nas mesmas condições. Se o extrato após purificação é armazenado deve-se evidenciar a estabilidade da micotoxina no mesmo após o armazenamento.
Os estudos de estabilidade podem ser realizados ao longo dos ensaios de rotina a partir da avaliação de solução padrão, de amostras já analisadas pelo laboratório, de material de referência, dentre outros.
¶ VI 4.2.8 Incerteza de Medição
A incerteza para os procedimentos analíticos que determinam micotoxinas em alimentos de origem vegetal ou animal, deve ser estimada seguindo os procedimentos citados no item II.12 deste Manual.
Ambas as metodologias aqui citadas, são usadas na estimação da incerteza dos procedimentos analíticos: metodologias denominadas de Bottom-Up e Top-Down.
Minimamente, para o procedimento Top-Down, deve-se levar em conta as contribuições dos seguintes componentes da incerteza:
- Precisão intermediária (reprodutibilidade interna);
- Veracidade / Recuperação (através do Bias ou Tendência);
- Curva de calibração.
Os procedimentos analíticos aplicados ao controle oficial devem produzir resultados cujas incertezas de medição padrão sejam inferiores à incerteza de medição padrão máxima calculada por meio da seguinte fórmula [30]:
Onde:
Uf = incerteza de medição padrão máxima (µg kg-1).
LD = limite de detecção do método (µg kg-1).
c = corresponde à concentração em causa (µg kg-1).
α = é um fator numérico cuja utilização depende de c.
Os valores de α a serem utilizados se encontram na Tabela 15 abaixo:
Tabela 15: Valores de α a serem utilizados.
|
c (µg kg-1) |
α |
|
≤ 50 |
0,2 |
|
51 a 500 |
0,18 |
|
501 a 1000 |
0,15 |
|
1001 a 10 000 |
0,12 |
|
> 10 000 |
0,1 |
¶ VI 4.3 Extensões de Escopo (Novos Analitos)
Quando se desejar incluir novas micotoxinas a um procedimento analítico previamente validado, o laboratório deve utilizar os mesmos parâmetros e critérios definidos anteriormente para garantir sua adequação (validação) após a inclusão.
Na introdução de uma nova micotoxina a seletividade deve ser avaliada. Caso não ocorram mudanças nas condições analíticas, repetir também as etapas de determinação do LQ, veracidade e precisão. Caso seja necessária alguma modificação analítica repetir o procedimento de validação integralmente.
Alternativamente, a validação de nova micotoxina pode ser realizada de forma concorrente, por intermédio do uso de amostras de controle de qualidade durante as análises de rotina, isto é, com cada lote de amostras da rotina um ou mais produtos da categoria são fortificados (n=3) na concentração do LQ, um nível maior - preferencialmente o nível requerido e um limite correspondente ao ponto da curva de calibração mais concentrado.
¶ VI 4.4 Extensões de Escopo (Novas Matrizes)
Quando a matriz a ser introduzida não pertencer a um grupo representativo de amostras Tabela 12 uma validação completa deve ser conduzida.
Quando a matriz pertencer a um determinado grupo representativo, a ampliação do escopo deverá ser feita a partir da avaliação de estudos de seletividade, precisão, veracidade e LQ; com no mínimo um dia de experimento conforme descrito neste Manual e deverá atender aos critérios de desempenho estabelecidos para a validação.
¶ VI 4.5 Alteração do Nível de Concentração Requerido
Caso seja necessário mudar o limite de referência (nível de interesse ou nível requerido) utilizado no processo de validação, e este estiver fora da faixa de trabalho da curva de calibração, deve-se repetir os experimentos de LQ, conforme o Item VI.4.2.5. – Limite de Detecção, Limite de Quantificação. Caso o novo limite esteja dentro da curva de calibração, avaliar o LD e LQ pelo menos uma vez.
¶ VI 4.6 Revalidação
Revalidação é o procedimento que deve ser adotado quando houver alteração nas condições originais da validação, que possam afetar os resultados de medição ou para avaliar o desempenho do procedimento analítico ao longo de um período conforme previsto pela Norma NBR/ISO/IEC 17025, item 5.4.5.2.
Dados de série histórica de controle intralaboratorial, participação em ensaios de proficiência, testes de homogeneidade de materiais, também podem ser utilizados para revalidar os procedimentos analíticos desde que atendam aos parâmetros exatidão e precisão.
Os parâmetros mínimos que devem ser avaliados na revalidação são: veracidade, precisão intermediária, LD, LQ e incerteza de medição do procedimento analítico.
¶ VI 4.7 Calibração Analítica e Recuperação (Rotina Analítica)
Todos os procedimentos adotados na rotina analítica deverão ser respaldados pelos estudos de validação do procedimento analítico.
¶ VI 4.7.1 Frequência Mínima para utilização da curva de Calibração na Rotina Analítica
Para as micotoxinas, cada batelada de amostras deve ser acompanhada de no mínimo uma curva de calibração com no mínimo cinco concentrações, sendo que pelo menos um ponto da curva de calibração contemple o LQ.
¶ VI 4.7.2 Determinação da Recuperação na Rotina Analítica
Os níveis de fortificação das amostras para determinação das recuperações das micotoxinas deverão estar na faixa da curva de calibração (e.g. no LR), ou dentro de um nível de relevância para as amostras analisadas. Esse nível de fortificação deve ser alternado ao longo do tempo. Na rotina, amostras com contaminação fora da faixa da curva de calibração deverão ser diluídas e reinjetadas.
É obrigatório um monitoramento contínuo dos procedimentos analíticos e dos resultados das medições obtidos na rotina laboratorial seja realizado com o objetivo de decidir se os resultados analíticos estão em conformidade com os requisitos estabelecidos, devendo ser realizado para todos os procedimentos analíticos utilizados na rotina. Assim, sempre que possível, em uma batelada contendo diferentes matrizes deve-se avaliar a recuperação de todas as micotoxinas analisadas em pelo menos uma matriz de cada grupo.
Recomenda-se plotar as recuperações obtidas em gráficos de controle (cartas de controle) preferencialmente por micotoxinas e grupo de matriz, considerando as faixas de contaminação. Caso seja observada uma tendência significativa ou sistemática de desvio de recuperação, procedimentos devem ser adotados no intuito de se identificar e corrigir as causas prováveis do desvio.
¶ VI 4.7.2.1 Critérios de Aceitação do Desempenho de Recuperação na Rotina Analítica
Os critérios de aceitabilidade, em termos de recuperação, para os resultados dos controles devem ser de acordo com aqueles definidos no item VI.4.2.4.4.
Se uma tendência significativa ocorrer na recuperação ou se resultados potencialmente inaceitáveis (desvio-padrão relativo maior que 20 %) forem obtidos, as causas devem ser investigadas.
¶ VI 4.8 Preparo de Soluções Padrão de Micotoxinas (que não são MRC)
As soluções padrão de micotoxinas devem ser preparadas e padronizadas de acordo com os procedimentos descritos na IN Nº 9 de 24/03/2000 do MAPA [54]. As soluções padrão utilizadas na rotina devem ser periodicamente monitoradas por repadronização, cabendo ao laboratório definir o período para repadronização das soluções padrão de micotoxinas.
As soluções padrão devem ser preparadas por injeção de solventes apropriados nos frascos (utilizando-se de seringa com agulha) para cada micotoxina, seguindo-se de retirada de frações das soluções concentradas para preparo das soluções estoque, evitando-se dessa forma a abertura dos frascos contendo padrão sólido e consequente dispersão das partículas.
As concentrações das soluções estoquem devem ser calculadas após determinação das absorbâncias das soluções por espectrofotometria UV. Previamente à determinação das absorbâncias das soluções padrão, o espectrofotômetro deve ser avaliado quanto ao seu desempenho por meio de soluções do padrão primário de dicromato de potássio segundo AOAC Internacional [4], ou pelo uso de padrão certificado por organismo nacional de metrologia (National Institute of Standart and Technology - NIST, LGC, Instituto Nacional de Metrologia - INMETRO, entre outros).
Para algumas micotoxinas não estão disponíveis valores de absortividade, desta forma devem ser preparadas por medição exata das massas seguida de transferências quantitativas das massas para frascos volumétricos calibrados e aferição criteriosa do volume ou utilizando micropipetas ou pipetas calibradas.
Na Tabela 16 estão apresentados os solventes apropriados para o preparo das soluções assim como os parâmetros a serem utilizados na padronização delas.
Tabela 16: Parâmetros para Padronização de Soluções de Micotoxinas.
|
Micotoxina |
Faixa de comp. de onda (nm) |
Massa Molecular (g/mol) |
Faixa de conc. ideal (μg/mL) |
Solvente Indicado |
Absortiv. Molar (∈) |
|
Aflatoxina B1 AFLAB1 |
330-370 |
312 |
8-10 |
Benzeno: Acetonitrila (98:2, v/v) |
19800 (350 nm) |
|
Tolueno: Acetonitrila (9:1, v/v) |
19300 (350 nm) |
||||
|
Metanol |
21500 (350 nm) |
||||
|
Acetonitrila |
20700 (350 nm) |
||||
|
Aflatoxina B2 AFLAB2 |
330-370 |
314 |
8-10 |
Benzeno: Acetonitrila (98:2, v/v) |
20900 (350 nm) |
|
Tolueno: Acetonitrila (9:1, v/v) |
21000 (350 nm) |
||||
|
Metanol |
21400 (350 nm) |
||||
|
Acetonitrila |
22500 (350 nm) |
||||
|
Aflatoxina G1 AFLAG1 |
330-370 |
328 |
8-10 |
Benzeno: Acetonitrila (98:2, v/v) |
17100 (350 nm) |
|
Tolueno: Acetonitrila (9:1, v/v) |
16400 (350 nm) |
||||
|
Metanol |
17700 (350 nm) |
||||
|
Acetonitrila |
17600 (350 nm) |
||||
|
Aflatoxina G2 AFLAG2 |
330-370 |
330 |
8-10 |
Benzeno: Acetonitrila (98:2, v/v) |
18200 (350nm) |
|
Tolueno: Acetonitrila (9:1, v/v) |
18300 (350 nm) |
||||
|
Metanol |
19200 (350 nm) |
||||
|
Acetonitrila |
18900 (350 nm) |
||||
|
Aflatoxina M1 AFLA M1 |
330-370 |
328 |
8-10 |
Benzeno: Acetonitrila (9:1, v/v) |
18000 (350 nm) |
|
Acetonitrila |
19000 (350 nm) |
||||
|
Clorofórmio |
1995 (350 nm) |
||||
|
Ocratoxina A OCRA ou OTA |
300-400 |
403 |
40 |
Benzeno:ácido Acético (99:1, v/v) |
5550 (330nm) |
|
Tolueno: Ácido Acético (99:1, v/v) |
5440 (333nm) |
||||
|
Zearalenona ZON |
280-380 |
318 |
50 |
Benzeno |
6060 (317nm) |
|
Metanol |
6000 (314 nm) 13900 (274 nm) 30000 (236 nm) |
||||
|
Desoxinivalenol DON |
200-400 |
296,3 |
50 |
Metanol |
7040 (219 nm) |
|
Acetonitrila |
6400 (208 nm) |
||||
|
Citreoviridina CTV |
200-400 |
402 |
5 |
Metanol |
44925 (383 nm) |
|
Etanol |
48000 (388 nm) |
Observações da tabela 16:
- O preparo das soluções na faixa ideal é desejado, mas não podendo ser atendido, deverá ser observado no espectro obtido se a altura do pico está aceitável.
- Soluções de DON em metanol podem sofrer degradação após 30 dias de preparo [4 e 57].
- Solução padrão de AFLA M1 é estável em acetonitrila por aproximadamente 1 mês [15].
¶ VI 4.9 Armazenamento de Padrões
Armazenar os padrões sólidos e suas soluções separadamente das amostras ou extratos de amostras, em frascos para padrão âmbar ou frascos protegidos da luz por papel alumínio, vedados com parafilme. A temperatura de armazenamento deve seguir a recomendação do fabricante.
¶ VI 4.10 Critérios de Aceitabilidade das Soluções Padrão de Micotoxinas
Recomenda-se que a diferença entre as concentrações determinadas na padronização anterior e a atual seja ≤ 3%
As soluções padrão de micotoxinas devem ser descartadas quando observadas alterações indicativas de deterioração como: turbidez, separação entre as fases, alteração de cor, entre outras.
A avaliação da pureza ou possível degradação das soluções padrão podem ser realizadas por técnicas de cromatografia em camada delgada e/ou cromatografia líquida.
¶ VI.5 Dioxinas e PCBs-dl (semelhantes a dioxinas)
Esta seção trata especificamente dos analitos descritos abaixo e Tabela 17:
- Dioxinas (dibenzo-para-dioxinas policloradas - PCDD) e furanos (dibenzofuranos policlorados - PCDF) contendo cloro nas posições 2,3,7,8;
- Difenilas policloradas semelhantes a dioxinas (PCB-dl).
Tabela 17: Analitos tratados na seção Dioxinas e PCBs
|
Dioxinas |
Furanos |
PCBs-dl* |
|
2,3,7,8-Tetraclorodibenzo-p-dioxina |
2,3,7,8-Tetraclorodibenzofurano |
PCB-81 |
|
1,2,3,7,8-Pentaclorodibenzo-p-dioxina |
1,2,3,7,8- Pentaclorodibenzofurano |
PCB-77 |
|
1,2,3,4,7,8-Hexaclorodibenzo-p-dioxina |
2,3,4,7,8-Pentaclorodibenzofurano |
PCB-126 |
|
1,2,3,6,7,8-Hexaclorodibenzo-p-dioxina |
1,2,3,4,7,8-Hexaclorodibenzofurano |
PCB-169 |
|
1,2,3,7,8,9-Hexaclorodibenzo-p-dioxina |
1,2,3,6,7,8-Hexaclorodibenzofurano |
PCB-123 |
|
1,2,3,4,6,7,8-Heptaclorodibenzo-p-dioxina |
2,3,4,6,7,8-Hexaclorodibenzofurano |
PCB-118 |
|
Octaclorodibenzo-p-dioxina |
1,2,3,7,8,9-Hexaclorodibenzofurano |
PCB-114 |
|
1,2,3,4,6,7,8-Heptaclorodibenzofurano |
PCB-105 |
|
|
1,2,3,4,7,8,9-Heptaclorodibenzofurano |
PCB-167 |
|
|
Octaclorodibenzofurano |
PCB-156 |
|
|
PCB-157 |
||
|
|
|
PCB-189 |
* Número IUPAC
Trata-se de um grupo de 17 PCDD/F e 12 PCBs-dl de elevada toxicidade. O TMC aplica-se ao somatório de dioxinas e furanos (PCDD/F) e/ou ao somatório total (PCDD/F + PCBs-dl). Para o cálculo do somatório, as concentrações de cada analito numa determinada amostra são multiplicadas pelos respectivos Fatores de Equivalência Tóxica (TEF), definidos pela Organização Mundial de Saúde, e somadas para resultar na concentração total de compostos sob a forma de dioxina, expressos em equivalentes de toxicidade (TEQ-OMS). Usualmente são utilizadas as unidades pgTEQ/g para alimentos e ngTEQ/kg para alimentos para animais.
¶ VI.5.1 Validação para métodos confirmatórios (Requisitos Mínimos e Critérios de Aceitação)
Os métodos de confirmação devem permitir identificação e quantificação inequívoca dos PCDD/F e PCB-dl em torno do limite de referência (TMC). Atualmente, tais métodos fazem uso de cromatografia a gás (CG) acoplada à espectrometria de massas de alta resolução (HRMS) e espectrometria de massas sequencial (MS/MS).
A Espectrometria de massas de alta resolução é definida como tendo resolução superior a 10.000 para todo o intervalo de massa a 10% da altura do pico [33].
Para métodos confirmatórios o uso de padrões internos é mandatório. No início da etapa analítica, devem ser adicionados padrões internos isotopicamente marcados com 13C. Deve-se utilizar um congênere marcado para cada PCDD/F tetra a octo-clorados (aceita-se a exceção de 1,2,3,7,8,9-HxCDD e OCDF), e a totalidade dos 12 congêneres marcados de PCBs-dl. A recuperação dos padrões internos deve ser avaliada, como ferramenta de controle de qualidade do método, conforme descrito em item específico deste manual.
Para análise em matrizes que contenham menos de 10% de gordura ou nas quais o limite de referência é aplicado por base de produto, a adição dos padrões internos deve ser realizada antes da etapa de extração.
Para análise em matrizes que contenham mais de 10% de gordura, ou nas quais o resultado é reportado por base de gordura, a adição do padrão interno pode ser realizada após a extração de gordura.
Deve ser efetuada uma validação adequada da eficácia da extração, dependendo da fase em que são introduzidos os padrões internos e de os resultados serem reportados em base de produto ou em base de gordura.
A validação de procedimentos analíticos para determinação quantitativa de PCDD/F e PCB-dl deverá ser conduzida observando-se os seguintes parâmetros de desempenho e os seus respectivos critérios de aceitação:
- Seletividade;
- Linearidade;
- Limite de Detecção;
- Limite de Quantificação;
- Recuperação/Veracidade;
- Recuperação do Padrão Interno
- Precisão (Repetitividade e Reprodutibilidade intralaboratorial);
- Incerteza de Medição.
V.5.1.1 Seletividade
Condições cromatográficas adequadas devem ser estabelecidas para garantir a separação das 17 PCDD/F (com cloro substituído nas posições 2,3,7,8) e dos 12 PCBs-dl, dos demais congêneres, assim como outros compostos clorados interferentes, como os éteres difenílicos clorados.
Os seguintes critérios devem ser avaliados:
- Separação cromatográfica que resulte em sobreposição, considerando altura do vale pico a pico, menor que 25%. Em especial para os pares de compostos 1,2,3,4,7,8-HxCDF /1,2,3,6,7,8-HxCDF, PCB-123/PCB-118. PCB156/PCB-157 e PCB169/PeCDD. 123478-HxCDD/123678-HxCDD e OCDD/OCDF.
- Recomenda-se o uso de padrões de checagem de resolução de coluna, os quais apresentam outros isômeros de PCDD/F. O critério descrito acima deve ser observado para todos os isômeros.
- A razão entre os tempos de retenção cromatográficos da substância a analisar e do padrão interno, isto é, o tempo de retenção relativo, deve corresponder ao da solução de calibração com uma tolerância de ± 0,5 %.
- Para CG-HRMS, a razão isotópica dos compostos deve obedecer à faixa apresentada na Tabela 18, que contempla variação máxima de 15% do valor teórico. Outros conjuntos de massa podem ser utilizados, desde que seja respeitada a variação máxima de 15% da razão teórica.
Tabela 18. Razão isotópica teórica e aceitável para as PCDD/F e PCB-dl de acordo com o número de cloros da molécula e com a natureza da razão m/z [60].
|
Número de átomos de cloro |
m/z |
Razão Teórica |
Limites aceitáveis |
|
|
Inferior |
Superior |
|||
|
4 |
M/(M+2) |
0.77 |
0.65 |
0.89 |
|
5 |
(M+2)/(M+4) |
1.55 |
1.32 |
1.78 |
|
6 |
(M+2)/(M+4) |
1.24 |
1.05 |
1.43 |
|
7 |
(M+2)/(M+4) |
1.05 |
0.88 |
1.20 |
|
8 |
(M+2)/(M+4) |
0.89 |
0.76 |
1.02 |
- Para GC-MS/MS: Monitoramento de, pelo menos, 2 íons precursores específicos, cada um com um íon-produto de transição específico correspondente, para cada analito marcado e não marcado. Tolerância máxima permitida das intensidades relativas de íons de ± 15 % para transições selecionadas de íons-produto em comparação com valores calculados ou medidos (média dos padrões de calibração) nas mesmas condições de MS/MS. A resolução para cada quadrupolo deve ser definida igual ou melhor do que a resolução de uma unidade de massa, de modo a minimizar eventuais interferências sobre os analitos de interesse.
¶ V.5.1.2 Linearidade
Construir uma curva de calibração com no mínimo cinco níveis de concentração, compreendendo preferencialmente os valores de LQ e quando possível o TMC, em três ou mais repetições por nível de calibração.
Alternativamente ao procedimento descrito no Item II.3, a avaliação da linearidade para análise de PCDD/F e PCBs-dl pode ser feita pela avaliação do Fator de Resposta Relativa - RRF (relative response factor). [60]. Esta alternativa é possível dada a obrigatoriedade do uso da técnica de diluição isotópica para métodos confirmatórios.
O RRF é calculado para cada ponto da curva de maneira independente, pela seguinte equação:
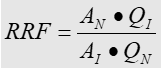
Onde,
An: resposta instrumental do analito nativo (não marcado) de interesse;
Ai: resposta instrumental do padrão interno marcado referente ao analito de interesse;
Qn: concentração ou quantidade do analito nativo (não marcado) de interesse;
Qi: concentração ou quantidade do padrão interno marcado referente ao analito de interesse;
Calcular o RRF para cada ponto da curva, o RRF médio e o coeficiente de variação entre os pontos da curva. Os resíduos (%) para cada ponto da curva, obtidos através do RRF médio devem também ser avaliados.
¶ V.5.1.2.1 Critérios de aceitabilidade
O valor do RRF deve se manter constante entre os pontos da curva, com coeficiente de variação menor que 20% [60].
Os resíduos devem ser plotados em gráfico de dispersão para avaliação de tendência ao longo da faixa de trabalho estudada.
¶ V.5.1.3 Limite de detecção
Limite de detecção do equipamento é definido como a quantidade injetada do analito que produz um sinal de pelo menos três vezes a razão sinal/ruído do equipamento para o íon menos intenso. Para determinar o limite de detecção, diluir o padrão até um nível de concentração detectável, calcular a quantidade injetada, considerando também os critérios de tempo de retenção e razão de íons.
¶ V.5.1.3.1 Critério de aceitabilidade
Devido à elevada toxicidade destes compostos o limite de detecção deve estar na ordem de pelo menos:
- fentogramas (10–15 g) para PCDD/F;
- picogramas (10–12 g) para os PCBs-dl não orto substituídos;
- nanogramas (10–9 g) para os PCBs-dl mono orto substituídos.
¶ V.5.1.4 Limite de quantificação
O limite de quantificação do procedimento analítico é definido como o nível mais baixo no qual foi demonstrado que os critérios de veracidade, precisão e seletividade foram atendidos.
Analisar amostra naturalmente contaminada ou fortificada no nível do limite de quantificação (n ≥ 6). Esta abordagem leva em consideração perdas ocorridas durante o procedimento analítico e deve ser usada preferencialmente. Em caso impossibilidade de uso de amostra na concentração desejada, para analitos específicos, a abordagem abaixo pode ser utilizada.
Considerar o LQ o ponto de concentração mais baixo da curva de calibração que apresenta um desvio padrão relativo aceitável (≤ 20%) e coerente em relação ao fator de resposta relativo (RRF) médio calculado para todos os pontos da curva de calibração.
¶ V.5.1.4.1 Critério de aceitabilidade
Para os congêneres individuais os critérios de veracidade, precisão e seletividade devem ser atendidos.
Como o nível de interesse para análise de PCDD/F e PCBs-dl usualmente é aplicado ao somatório (TEQ) no nível upperbound (também chamado de limite superior de concentração), este conceito também deverá ser aplicado na avaliação do limite de quantificação do método. Especialmente para métodos de monitoramento, o limite de quantificação deverá ser tão baixo quanto possível. Preferencialmente o método deverá ter limite de quantificação de cerca de 1/5 do nível de interesse [31] sendo este considerado o limite de quantificação alvo. Para determinadas matrizes, especialmente nas quais o nível de interesse é baixo (≤ 1 pgT EQ/g) pode ser aceitável até cerca de 1/3 do nível de interesse.
Para avaliação se o método é adequado ao propósito de uso, a diferença entre os níveis upperbound e lowerbound para amostras que superem o TMC ou nível de interesse deve ser menor que 20 %.
O nível upperbound preconiza que, no somatório das PCDD/F e/ou PCBs-dl, a contribuição de cada congênere não quantificado seja igual ao limite de quantificação; no nível lowerbound, a contribuição de cada congênere não quantificado é igual a zero.
¶ V.5.1.5 Recuperação/Veracidade e Precisão (Repetitividade e Reprodutibilidade Intralaboratorial)
Analisar, no mínimo, seis replicatas de amostras em 3 níveis de concentração, preferencialmente contemplando o 0,5 vezes o limite de referência (TMC); 1,0 vez do TMC e 2,0 vezes o TMC requerido para os compostos. O primeiro nível pode ser substituído pelo LQ do método, quando aplicável.
Repetir o procedimento acima, no mínimo em mais duas ocasiões (em condições de reprodutibilidade intralaboratorial).
Adicionalmente recomenda-se o uso de MR ou MRC para avaliação da veracidade.
Calcular as porcentagens de recuperação e coeficientes de variação (CV) das replicatas por nível de concentração.
¶ V.5.1.5.1 Critérios de Aceitabilidade
Como critério para a veracidade aceita-se -20 a +20 % de variação em relação ao valor alvo, para cada nível avaliado.
A precisão em termos de reprodutibilidade intralaboratorial (CVR) deve ser menor que 15%.
A precisão em termos de repetitividade (CVr) deve ser menor que 15%.
¶ V.5.1.6 Recuperação do padrão interno
Antes da análise em equipamento GC, devem ser adicionados em cada amostra um ou mais padrões de recuperação (também denominados padrões de seringa) para avaliação da recuperação do padrão interno (adicionado no início da extração).
O controle desta recuperação é feito conforme a equação:
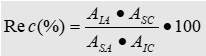
Onde,
- RecPI = recuperação do padrão interno
- AIA = área do padrão interno na amostra
- ASC = área do padrão de recuperação na solução de calibração
- ASA = área do padrão de recuperação na amostra
- AIC = área do padrão interno na solução de calibração
Caso a quantidade de padrões adicionados às amostras e à solução de calibração sejam diferentes, estas quantidades devem ser consideradas.
¶ V.5.1.6.1 Critérios de Aceitabilidade
A faixa de recuperação alvo para os padrões internos é de 60 a 120 %. Recuperações fora desta faixa para congêneres individuais, em particular hepta e octa-CDD/F e PCBs-dl são aceitáveis, na condição de que suas contribuições para os somatórios de PCDD/F e PCDD/F + PCB-dl não excedam a 10 % do valor total calculado. Valores de recuperação de padrão interno para os demais compostos fora dessa faixa podem ser aceitos desde que seja demonstrada a acurácia na quantificação do composto nativo correspondente nos ensaios de validação e nos controles de rotina. Valores de recuperação menores que 30% indicam perda excessiva no procedimento analítico e devem ser evitados.
¶ V.5.1.7 Incerteza de Medição
A incerteza de medição deve ser estimada segundo procedimento estabelecido no Item II.12, deste Manual.
A incerteza de medição deve ser estimada para os analitos de interesse, assim como para o somatório de dioxinas e furanos (PCDD/F TEQ) e para o somatório total (PCDD/F + PCBs-dl TEQ), aos quais se aplicam o TMC. Para os somatórios o cálculo deve preferencialmente considerar a raiz quadrada da soma dos quadrados dos valores de incerteza padrão individuais, porém outras abordagens descritas podem ser utilizadas [28].
V.5.1.7.1 Critério de aceitabilidade
Os procedimentos analíticos aplicados ao controle oficial devem produzir resultados cujas incertezas de medição padrão sejam inferiores à incerteza de medição padrão máxima calculada por meio da seguinte equação:
Onde:
uf é a incerteza medida padrão máxima;
LD é o limite de detecção do método;
C é a concentração de interesse;
α é o fator numérico cuja utilização depende do valor de C (para métodos de PCDD/F e PCBs-dl em matrizes de alimentos, α = 0,2).
¶ V.5.1.8 Procedimento para extensão de escopo
Novas matrizes a serem incluídas num método previamente validado necessitam de verificação de desempenho, utilizando os mesmos parâmetros e critérios definidos anteriormente.
Os critérios de seletividade e linearidade e recuperação do padrão interno devem ser atendidos para a nova matriz.
Para avaliação da precisão e da veracidade/recuperação do método, os ensaios de amostras fortificadas devem ser realizados no mínimo em duplicata, em três níveis e devem ser repetidos em pelo menos mais dois dias, até que se obtenham no mínimo 6 replicatas por nível. Deve ser incluída a avaliação do LQ para a nova matriz.
Para matrizes similares, que passem pelo mesmo procedimento de extração e purificação, como gordura animal de espécies diferentes; ou alimentos para animais de diferentes categorias, pode ser adotado procedimento de extensão de escopo multi matrizes, desde que compreenda o descrito no parágrafo anterior para cada matriz.
Os valores de recuperação média e de coeficiente de variação obtidos devem estar dentro dos intervalos aceitáveis utilizados na validação do método para o analito ou a matriz original.
¶ V.5.2 Validação para métodos de triagem
¶ V.5.2.1 Requisitos gerais para métodos de triagem
Os métodos de triagem devem basear-se em ensaios bioanalíticos ou em GC-MS/MS.
Os métodos de triagem apenas podem ser aplicados para o controle de uma determinada matriz se forem suficientemente sensíveis, considerando o nível de interesse ou TMC. Estes métodos não são adequados para a finalidade de avaliação dos níveis de base (background), estimativa da ingestão ou avaliação de tendências ao longo do tempo.
O resultado emitido pelo método de triagem deve ser expresso como conforme ou suspeito de ser não conforme (negativo / positivo).
Os laboratórios que aplicam métodos de triagem devem obrigatoriamente ter disponível o método confirmatório. As amostras processadas em método de triagem e consideradas suspeitas de serem não conformes em relação ao TMC devem ser reanalisadas utilizando um método confirmatório.
A aplicação do método deve objetivar evitar resultados falsos conformes (falso negativos). É necessária a verificação de desempenho durante a análise de rotina, por procedimentos de garantia da qualidade analítica e validação contínua do método. Deve existir um programa contínuo para a verificação dos resultados conformes.
¶ V.5.2.1.1 Valor de corte
Deve ser estabelecido um valor de corte para a decisão sobre a conformidade da amostra em relação ao nível de interesse ou TMC fixados para PCDD/F, ou para a soma de PCDD/F e PCB-dl. Quando o método não contempla a separação das duas frações, deve-se considerar o menor nível de interesse ou TMC, ou seja, o nível aplicado para PCDD/F apenas.
¶ V.5.2.1.1.1 Estimativa do valor de corte
O valor de corte pode ser estimado como:
- 2/3 do limite máximo permitido. Neste nível devem ser atendidos os critérios de taxa de resultados falsos-conformes menor que 5% e taxa de resultados falsos-não-conformes adequada (Tabela 3), ou;
- Realizar múltiplas análises (n ≥ 6), em condições de reprodutibilidade intralaboratorial, de amostra em nível de concentração próximo ao limite de decisão esperado do método confirmatório e estimar o valor de corte de acordo com a equação abaixo:
Valor de corte = R – 1,64 * SDR
Onde:
R é o resultado (concentração para o somatório) médio encontrado das replicatas;
SDR é o valor do desvio padrão medido em condições de reprodutibilidade interna
Outras abordagens descritas em [31] ou literatura específica podem ser utilizadas.
Em caso de análise de amostras provenientes de incidentes de contaminação, os valores de corte podem ser reavaliados, refletindo a matriz e os perfis de congêneres específicos para cada incidente.
¶ V.5.2.1.2 Critérios para taxa de falso conforme e precisão
Os critérios a serem atendidos para métodos de triagem estão descritos na Tabela 19.
Não há critério para a taxa de falso não conforme (falso positivo). Esta deve ser suficientemente baixa para tornar benéfico o uso do método como instrumento de triagem.
Tabela 19: Critérios para avaliação de métodos de triagem para PCDD/F e PCBs-dl [31].
|
Parâmetro |
Valor máximo aceitável (%) |
|
Taxa de falso conforme |
<5 |
|
Veracidade |
- |
|
Precisão (DPRr) |
<20 |
|
Precisão (DPRR) |
<25 |
¶ V.5.2.1.3 Avaliação de amostras conformes
2 a 10% das amostras conformes em métodos de triagem devem ser submetidas ao método confirmatório para avaliação contínua da taxa de falso conforme. Este percentual deve ser aplicado para cada matriz analisada.
¶ V.5.2.2 Requisitos específicos para métodos de triagem por GC-MS/MS
Para métodos de triagem que utilizam GC-MS/MS, parâmetros adicionais aos descritos no item anterior devem ser avaliados.
¶ V.5.2.2.1 Ensaios de validação
Analisar, no mínimo, 07 replicatas de amostras em pelo menos 02 níveis de concentração, contemplando amostras abaixo do valor de corte e no valor de corte. Repetir o procedimento acima, no mínimo em mais duas ocasiões, até que sejam obtidos pelo menos 21 replicatas por nível.
¶ V.5.2.2.2 Uso de padrão interno
O uso de padrões internos é mandatório. No início da etapa analítica, devem ser adicionados padrões internos de marcados com 13C, com pelo menos um congênere para cada grupo homólogo de PCDD/F e PCBs-dl.
Antes da análise em equipamento GC, devem ser adicionados em cada amostra um ou mais padrões de recuperação (também denominados padrões de seringa) para avaliação da recuperação do padrão interno (adicionado no início da extração).
¶ V.5.2.2.2.1 Critério
Para métodos de triagem por GC-MS, as recuperações do padrão interno devem situar-se na faixa de 30 a 140 %.
¶ V.5.2.3 Requisitos específicos para métodos de triagem por bioensaio
Para métodos que utilizam bioensaio, parâmetros adicionais aos descritos no item 1.2.1 devem ser avaliados.
Os métodos bioanalíticos são métodos baseados na utilização de princípios biológicos, como ensaios com células, ensaios com receptores ou imunoensaios. Não fornecem resultados para cada congênere individual, mas apenas uma indicação do somatório (TEQ). A expressão do resultado do bioensaio deve ser feita em equivalentes bioanalíticos (BEQ).
As soluções padrão e os extratos de amostras devem ser testados em duplicata. A variação da resposta para um mesmo extrato ou padrão deve obedecer ao critério de CV < 15 %.
¶ V.5.2.3.1 Ensaios de validação
Analisar, no mínimo, 07 replicatas de amostras em pelo menos 03 níveis de concentração, contemplando amostras abaixo do valor de corte; no valor de corte; e acima do limite de referência ou TMC. Repetir o procedimento acima, no mínimo em mais duas ocasiões, até que sejam obtidos pelo menos 21 replicatas por nível.
O uso de amostras naturalmente contaminadas é essencial para abranger os perfis de congêneres mais frequentes, que representem diferentes fontes de contaminação. Excepcionalmente na ausência destas, podem ser utilizadas amostras fortificadas.
¶ V.5.2.3.2 Calibração
¶ V.5.2.3.2.1 Calibração com curva de padrão em solvente
Os teores nas amostras podem ser estimados por comparação da resposta ao teste com uma curva de calibração da TCDD ou de uma mistura-padrão de PCDD/F e PCBs-dl.
As curvas de calibração devem conter oito a 12 concentrações (em triplicata), com concentrações suficientes na parte inferior da curva, com especial atenção à qualidade de ajuste da curva na faixa de trabalho.
¶ V.5.2.3.2.2 Calibração com uso de material de referência matrizado
De forma alternativa, pode utilizar-se uma curva de calibração preparada a partir de, pelo menos, quatro amostras de referência: um ensaio em branco da matriz e três amostras de referência com 0,5 vezes, uma vez e duas vezes o TMC ou nível de interesse.
Neste caso, o valor de corte correspondente a dois terços do TMC ou nível de interesse pode ser calculado diretamente pela curva de calibração.
¶ V.5.2.3.3 Controle da recuperação
A eficiência da extração e perda de compostos durante as etapas de purificação do extrato deve ser verificada durante a validação. Uma amostra de concentração conhecida, contendo diferentes congêneres deve ser analisada pelo menos em triplicata por método confirmatório. A veracidade (recuperação em relação ao valor alvo) deve situar-se entre 60% e 120%, em especial para congéneres que contribuam mais de 10 % para o valor do somatório TEQ.
¶ V.5.2.3.4 Recuperação aparente do bioensaio
Para métodos bioanalíticos, deve ser determinada a recuperação aparente do bioensaio. Deve ser calculada a partir de material de referência com perfil de congêneres representativos próximos ao TMC ou nível de interesse. É expressa em percentagem do valor BEQ encontrado em comparação com o valor teórico em TEQ. O resultado deve situar-se conforme a Tabela 20.
Este valor, chamado de recuperação aparente do bioensaio, deve ser utilizado para correção do resultado. Este procedimento corrige:
- Perdas durante a extração;
- Diferenças de respostas analíticas por GC-MS e bioensaio.
Tabela 20: Critérios para recuperação aparente do bioensaio [31].
|
Grupo |
Recuperação aparente (%) |
|
PCDD/F + PCBs-dl (total) |
30 – 130 |
|
PCDD/F |
50 – 130 |
|
PCBs-dl |
20 – 60 |
Caso seja utilizada curva com material de referência matrizado, e não curva em solvente, elimina-se a necessidade de correção em função do ensaio em branco e da recuperação aparente. Neste caso as propriedades da matriz do material de referência devem corresponder às das amostras com teor desconhecido.
¶ V.5.2.4 Procedimento para extensão de escopo
Novas matrizes a serem incluídas num método previamente validado necessitam de verificação de desempenho, utilizando os mesmos parâmetros e critérios definidos anteriormente.
Os critérios descritos acima devem ser atendidos para a nova matriz.
Analisar, no mínimo, 10 replicatas de amostras em pelo menos 02 níveis de concentração, contemplando amostras abaixo do valor de corte e no valor de corte. Repetir o procedimento acima, no mínimo em mais uma ocasião, até que sejam obtidos pelo menos 20 replicatas por nível.
¶ VI.6 Hidrocarbonetos Policíclicos Aromáticos (HPAs)
A validação de procedimentos analíticos para determinação quantitativa de HPAs deverá ser conduzida observando-se os seguintes parâmetros de desempenho e os seus respectivos critérios de aceitação:
- Seletividade;
- Linearidade;
- Efeito de matriz;
- Limite de Detecção;
- Limite de Quantificação;
- Recuperação/Veracidade;
- Precisão (Repetitividade e Reprodutibilidade interna); e
- Incerteza de Medição.
¶ VI.6.1 Validação para métodos confirmatórios (Requisitos Mínimos e Critérios de Aceitação)
¶ VI.6.1.1 Seletividade
Analisar, sempre que disponível, um número apropriado de amostras brancas e averiguar (n ≥ 20) a presença de qualquer interferente (pico cromatográfico, sinal, traço de íon) na região de interesse que se espera a eluição do analito.
Quando aplicável, fortificar amostras em branco representativas numa concentração relevante com substâncias que possam interferir na identificação e/ou na quantificação da substância a analisar.
¶ VI.6.1.1.2 Critérios de Aceitabilidade da Seletividade
O sinal interferente deve ser ≤ 30% do sinal na concentração do Menor Nível Calibrado.
¶ VI.6.1.2 Efeito de Matriz
Aplicar os procedimentos de avaliação do Efeito Matriz conforme descrito na Item II.4 deste manual.
Caso a ocorrência de EM já seja conhecida e a necessidade de se trabalhar com CCM ou CCF já tenha sido identificada, não há a necessidade de realizar o estudo de EM.
O estudo de EM deve obrigatoriamente ser realizado nos casos em que for utilizada a CCS para quantificar as amostras.
¶ VI.6.1.2.1 Critérios de Aceitabilidade
A ocorrência de Efeito Matriz indica que o efeito de matriz precisa ser considerado ao preparar a curva de calibração, logo, curvas de calibração em solvente (CCS) não poderá ser utilizada.
No caso de presença de efeito matriz é necessário o uso da CCM ou da CCF para quantificar as amostras.
¶ VI.6.1.3 Linearidade
Os procedimentos relacionados à curva de calibração devem ser realizados conforme descrito no Item VI.3.2.
¶ VI.6.1.3.1 Critérios de aceitabilidade da Linearidade
Adota-se o critério de valores de R2 iguais ou maiores do que 0,95 para curvas preparadas em matriz (CCM ou CCF) e 0,97 para curvas preparadas em solvente (CCS).
¶ VI.6.1.4 Limite de detecção
O LD deve ser calculado pela seguinte abordagem [29]:
Selecionar 10 amostras brancas, de preferência representativas de diferentes tipos de matrizes.
As mesmas amostras brancas analisadas para o estudo de seletividade/especificidade podem ser usadas para a determinação do LD.
Analisar as amostras em condições de repetibilidade, juntamente com uma curva de calibração do tipo definido para toda a validação.
Calcular os parâmetros da curva analítica.
Determinar o desvio padrão do sinal das amostras brancas.
Determinar o LD usando a seguinte equação:
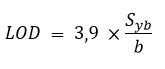
Onde:
LOD = limite de detecção
Syb = desvio-padrão do sinal do branco (pseudo-brancos)
b = inclinação da curva de calibração
¶ VI.6.1.4.1 Critério de aceitabilidade
O limite de detecção deve ser ≤ 0,30 µg/Kg para cada analito, especialmente para bBenzo(a)pireno, benzo(a)antraceno, benzo(b)fluoranteno e criseno.
¶ VI.6.1.5 Limite de quantificação
O LQ pode ser calculado pela seguinte abordagem [29]:
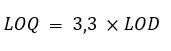
Onde:
LOD = limite de detecção
LOQ – limite de quantificação
¶ VI.6.1.5.1 Critério de aceitabilidade
O limite de quantificação deve ser ≤ 0,90 µg/Kg para benzo(a)pireno, benz(a)antraceno, benzo(b)fluoranteno e criseno.
¶ VI.6.1.6 Recuperação/Veracidade e Precisão (Repetitividade e Reprodutibilidade Intralaboratorial)
A analisar, no mínimo, seis replicatas de amostras em 3 níveis de concentração, preferencialmente contemplando o 0,5 vezes o limite de referência (TMC); 1,0 vez do TMC e 2,0 vezes o TMC requerido para os compostos. O primeiro nível pode ser substituído pelo LQ do método, quando aplicável.
Repetir o procedimento acima, no mínimo em mais duas ocasiões (em condições de reprodutibilidade intralaboratorial).
Adicionalmente recomenda-se o uso de MR ou MRC para avaliação da veracidade.
Calcular as porcentagens de recuperação e coeficientes de variação (CV) das replicatas por nível de concentração.
¶ VI.6.1.6.1 Critérios de Aceitabilidade
Como critério para a veracidade aceita-se recuperações de 50 a 120% em relação ao valor alvo, para cada nível avaliado.
A precisão em termos de reprodutibilidade intralaboratorial deve ter valor de HORRATR < 2.
A precisão em termos de repetitividade deve ter valor de HORRATr < 2.
O cálculo do valor de HORRAT é descrito no Item VI.3.6 Precisão.
¶ VI.6.1.7 Incerteza de Medição
A incerteza para os procedimentos analíticos que determinam HPAs deve ser estimada seguindo os procedimentos descritos no Item II.12 deste manual.
¶ VI.6.1.7.1 Critério de aceitabilidade
Os procedimentos analíticos aplicados ao controle oficial devem produzir resultados cujas incertezas de medição padrão sejam inferiores à incerteza de medição padrão máxima calculada por meio da seguinte fórmula [30]:
Onde:
Uf = incerteza de medição padrão máxima (µg kg-1).
LD = limite de detecção do método (µg kg-1).
c = corresponde à concentração em causa (µg kg-1).
α = é um fator numérico cuja utilização depende de c.
Os valores de α a serem utilizados se encontram na Tabela 21 abaixo:
Tabela 21: valores de α a serem utilizados.
|
c (µg kg-1) |
α |
|
≤ 50 |
0,2 |
|
51 a 500 |
0,18 |
|
501 a 1000 |
0,15 |
|
1001 a 10 000 |
0,12 |
|
> 10 000 |
0,1 |
¶ VI.6.1.8 Procedimento para extensão de escopo
O estudo de extensão de escopo será baseado em um lote de análise contendo todas as substâncias de interesse. O número de amostras do lote será de pelo menos 6 amostras por nível de validação fortificadas em três níveis diferentes, conforme descrito anteriormente, e a avaliação da curva de calibração.
Os critérios descritos para a validação original são aplicáveis.
¶ 5. Base legal e documentos de referência
¶ 5.1 Base legal
Não aplicável.
¶ 5.2 Documentos de referência
- ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS (ABNT). ABNT ISO GUIA 35: materiais de referência — guia para caracterização e avaliação da homogeneidade e estabilidade. 2. ed. 2020.
- ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS (ABNT). ABNT NBR ISO 17034: Requisitos gerais para competência de produtores de material de referência. 2017.
- ASSOCIAÇÃO BRASILEIRA DE NORMAS TÉCNICAS (ABNT). ABNT NBR ISO/IEC 17025:2017: Requisitos gerais para a competência de laboratórios de ensaio e calibração. 2017.
- ASSOCIATION OF OFFICIAL ANALYTICAL CHEMISTS (AOAC International). Official Methods of Analysis of AOAC International, 22nd ed.; Latimer, G. W., Ed.; Chapter 49.2.02; AOAC International: 2023.
- BARROS NETO, B.; SCARMINIO, I.S.; BRUNS, R. E. Como fazer experimentos. 3. ed. Campinas, SP: Editora Unicamp, 2007. ISBN 978-85-268-0753-2.
- BOYD, B.; BETHEM, R.; BASIC, C.; Trace quantitative analysis by mass spectrometry. Wiley, 2008, 752 p., ISBN 978-0-470-05771-1
- CODEX ALIMENTARIUS COMISSION. Draft. REP23/MAS-Appendix III. Information document procedures for the estimation of measurement uncertainty (for publication on the Codex website)
- CODEX ALIMENTARIUS COMISSION. Guideline CAC/GL 41-1993. PORTION OF COMMODITIES TO WHICH MAXIMUM RESIDUE LIMITS APPLY AND WHICH IS ANALYZED
- CODEX ALIMENTARIUS COMISSION. Guideline CXG 40-1993 Guidelines on good laboratory practice in pesticide residue analysis. 2010
- CODEX ALIMENTARIUS COMISSION. Guideline CXG 54-2004 Guidelines on mensurement of Uncertainty. 2021
- CODEX ALIMENTARIUS COMISSION. Guideline CXG 59-2006 Guidelines on Estimation of Uncertainty of Results. 2011
- CODEX ALIMENTARIUS COMISSION. Guideline CXG 72-2009 Guidelines on analytical terminology. 2009
- CODEX ALIMENTARIUS COMISSION. Guideline CXG 90-2017 Guidelines on performance criteria for methods of analysis for the determination of pesticide residues in food and feed. 2017
- DASS, C. Fundamentals Of Contemporary Mass Spectrometry - Wiley, 2010, 608 p. ISBN 978-0471682295
- DRAGACCI, S.; GROSSO, F. Immunoaffinity column clean-up with liquid chromatography for determination of aflatoxin M1 in liquid milk: collaborative study. Journal of AOAC International, v. 84, n. 2, 2001.
- ELLISON, S.L.R.; WILLIAMS, A. Eurachem/CITAC guide: Quantifying Uncertainty in Analytical Measurement, Third edition, (2012) ISBN 978-0-948926-30-3. Available from www.eurachem.org.
- EUROPEAN COMMISSION (EC). Commission Directive 2002/63/EC of 11 July 2002 establishing Community methods of sampling for the official control of pesticide residues in and on products of plant and animal origin and repealing Directive 79/700/EEC. Official Journal of the European Communities, 2002.
- EUROPEAN COMMISSION (EC). Commission Regulation (EU) 2017/771 of 3 May 2017 amending Regulation (EC) No 152/2009 as regards the methods for the determination of the levels of dioxins and polychlorinated biphenyls
- EUROPEAN COMMISSION (EC). Document SANTE 11312/2021 v2. Analytical quality control and method validation procedures for pesticide residues analysis in food and feed.
- EUROPEAN COMMISSION (EC). Document SANTE/12089/2016. Guidance Document on Identification of Mycotoxins in Food and Feed.
- EUROPEAN COMMISSION (EC). EURL Guidance Document on Confirmation Method Validation. Versão 1.1, 2021
- EUROPEAN COMMISSION (EC). EURL Guidance Document on Screening Method Validation. Versão 1.1 2023
- EUROPEAN COMMISSION (EC). EURL Guidance Document on Screening Method Validation. Versão 1.1, 2023
- EUROPEAN COMMISSION (EC). EURL Guidance document on the extension of quantitative confirmation methods. Versão 1.0, 2021
- EUROPEAN COMMISSION (EC). EURL guidance document on the quality control during routine analysis (ongoing method performance verification). Versão 1.2, 2023
- EUROPEAN COMMISSION (EC). EURL guidance on minimum method performance requirements (MMPR) for specific pharmacologically active substances in specific animal matrices. Versão 2, 2022
- EUROPEAN COMMISSION (EC). EURL-MP-guidance doc_003 (version 1.4) Guidance document on performance criteria for methods of analysis for mycotoxins and plant toxins in food and feed (draft 17 May 2024), Wageningen University and Research
- EUROPEAN COMMISSION (EC). Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry. Working Group for Measurement Uncertainty in PCDD/F and PCB Analysis - EURL 2017
- EUROPEAN COMMISSION (EC). Joint Research Centre, Robouch, P., Stroka, J., Haedrich, J., Schaechtele, A. et al., Guidance document on the estimation of LOD and LOQ for measurements in the field of contaminants in feed and food, Publications Office. 2016. ISBN 978-92-79-61768-3
- EUROPEAN COMMISSION (EC). Regulamento (CE) nº 333/2007 da Comissão, de 28 de Março de 2007, que estabelece métodos de amostragem e de análise para o controle oficial dos teores de chumbo, cádmio, mercúrio, estanho na forma inorgânica, 3-MCPD e benzo(a)pireno nos gêneros alimentícios.
- EUROPEAN COMMISSION (EC). Regulamento (UE) 2017/644 DA COMISSÃO de 5 de abril de 2017 que estabelece métodos de amostragem e análise para o controlo dos teores de dioxinas, PCB sob a forma de dioxina e PCB não semelhantes a dioxinas em determinados géneros alimentícios e que revoga o Regulamento (UE) nº 589/2014.
- EUROPEAN COMMISSION (EC). Regulamento (UE) N.o 752/2014 DA COMISSÃO de 24 de junho de 2014 que substitui o anexo I do Regulamento (CE) n.o 396/2005 do Parlamento Europeu e do Conselho.
- EUROPEAN COMMISSION (EC). Regulamento de Execução (UE) 2021/808 DA COMISSÃO de 22 de março de 2021. Relativo ao desempenho dos métodos analíticos para os resíduos de substâncias farmacologicamente ativas utilizadas em animais produtores de géneros alimentícios e à interpretação dos resultados, bem como aos métodos a utilizar na amostragem, e que revoga as Decisões 2002/657/CE e 98/179/CE
- EUROPEAN COMMISSION (EC). Regulamento de Execução (UE) 2023/2782 DA COMISSÃO de 14 de dezembro de 2023. Que estabelece os métodos de amostragem e de análise para controle dos teores de micotoxinas nos gêneros alimentícios e que revoga o Regulamento (CE) nº 401/2006.
- FUNK, W., DAMMANN, V. DONNEVERT, G. Quality Assurance in Analytical Chemistry, 2nd Ed., Weinheim, Germany. WILEY-VCH Verlag GmbH & Co. KGaA, 2007, 277 p. ISBN 978-3-527-31114-9
- HOFFMANN, E.; STROOBANT V. Mass Spectrometry - Principles and Applns 3rd ed - Wiley, 2007, 512 p., ISBN 978-0-470-03311-1
- HORWITZ, W. Evaluation of analytical methods used for regulation of foods and drugs. Analytical chemistry, v. 54, n. 1, p. 67-76, 1982.
- INSTITUTO NACIONAL DE METROLOGIA, NORMALIZAÇÃO E QUALIDADE INDUSTRIAL (INMETRO). Guia para a Expressão da Incerteza de Medição, 3. Ed. Edição brasileira do Guide to the Expression of Uncertainty in Measurement. rev. Rio de Janeiro: ABNT/INMETRO, ago. 2003.
- INSTITUTO NACIONAL DE METROLOGIA, NORMALIZAÇÃO E QUALIDADE INDUSTRIAL (INMETRO). Orientação sobre validação de métodos analíticos: documento de caráter orientativo. DOQ-CGCRE-008, Revisão 09 – JUN/2020. 2020.
- INSTITUTO NACIONAL DE METROLOGIA, NORMALIZAÇÃO E QUALIDADE INDUSTRIAL (INMETRO). Vocabulário Internacional de Metrologia: Conceitos fundamentais e gerais e termos associados (VIM 2012). Rio de Janeiro: Instituído pela Portaria n.º 232, de 08 de maio de 2012. 2012.
- INTERNACIONAL STANDARDS ORGANIZATION (ISO). Accuracy (trueness and precision) of measurement methods and results part 3: Intermediate precision and alternative designs for collaborative studies. Geneva, 2a Edição, 2023. 57p. ISO 5725-3;
- INTERNACIONAL STANDARDS ORGANIZATION (ISO). Accuracy (trueness and precision) of method measurements and results part 2: basic method for the determination of the repeatability and reproducibility of a standard measurement method. Geneva, 1994. 42 p. ISO 5725-2
- INTERNACIONAL STANDARDS ORGANIZATION (ISO). Capability of detection – part 1: terms and definitions. Geneva, 1997. 10 p. ISO 11843-1.
- INTERNACIONAL STANDARDS ORGANIZATION (ISO). Capability of detection – part 2: methodology in the linear calibration Case. Geneva, 2000. 24 p. ISO 11843-2.
- INTERNACIONAL STANDARDS ORGANIZATION (ISO). Guidance for the use of repeatability, reproducibility and trueness estimates in measurement uncertainty evaluation, Genova, 2017. 38p. ISO 21748:2017
- INTERNACIONAL UNION OF PURE AND APLLIED CHEMISTRY ORGANIZATION (IUPAC)/ISO/AOAC. Harmonized Guidelines for Single-Laboratory Validation of Methods of Analysis. Pure and Applied Chemistry. Thompson, M., Ellison, S.L.R. & Wood, R. 2002
- JOINT COMMITTEE FOR GUIDES IN METROLOGY (JCGM). Documento 100:2008. GUM 1995 with minor corrections Evaluation of measurement data — Guide to the expression of uncertainty in measurement. 2008
- JOINT COMMITTEE FOR GUIDES IN METROLOGY (JCGM). Documento 101:2008 Evaluation of measurement data — Supplement 1 to the “Guide to the expression of uncertainty in measurement” — Propagation of distributions using a Monte Carlo method
- MAGNUSSON, B.; ÖRNEMARK, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, (2nd ed. 2014). ISBN 9789187461590. Available from www.eurachem.org
- MAGNUSSON, B; NÄYKKI, T.; HOVIND, H.; KRYSELL, M.; SAHLIN, E. Handbook for calculation of measurement uncertainty in environmental laboratories, Nordtest Report TR 537 (ed. 4) 2017. Available from www.nordtest.info.
- MASSART, D. L. et al. Handbook of Chemometrics and Qualimetrics Part A (Data Handling in Science and Technology, Volume 20A). Elsevier, 1997. ISBN 978-0444897244
- MEIER, C.P.; ZUND E.R. Statistical Methods in Analytical Chemistry. 2nd ed. New York: Wiley Intersciences Publications, John Willey & Sons, 2000, p. 424. ISBN 0-471-29363-6.
- MILLER, J. N.; MILLER, J. C. Statistics and Chemometrics for Analytical Chemistry. 5th ed. Harlow, England: Pearson Prentice Hall, 2005. 268 p. ISBN-10: 0-13-129192-0
- MINISTÉRIO DA AGRICULTURA E PECUÁRIA (MAPA). Instrução Normativa Nº 9 de 24/03/2000. Aprova os métodos analíticos de referência de micotoxinas em produtos e subprodutos e derivados de origem vegetal.
- NIESSEN, W.M.A. Liquid Chromatography-Mass Spectrometry (Chromatographic Science Series) 3th ed - CRC Press, 2006, 632 p. ISBN 978-0824740825
- OLIVARES, I.R.B. Requisitos e Funcionamento de um Sistema de Gestão da Qualidade em Laboratórios 5a Edição, Editora Átomo, 2023, 190 p. ISBN 978-6587322223
- SHEPHERD, M., GILBERT, J., Long-Term Storage stability of Deoxynivalenol Standard Reference Solutions. Journal Agriculture of Food Chemistry, v.36, p. 305-308. 1988.
- SKOOG, D. A.; WEST, D.M.; HOLLER, J.F.; CROUCHM, S.R.; Fundamentos de Química Analítica: Tradução da 10ª edição norte-americana. Cengage Learning. 2023, 1088 p. ISBN 978-6555584240
- SOUZA, S. V. C. de. Procedimento para Validação Intralaboratorial de Métodos de Ensaio: delineamento e aplicabilidade em análise de alimentos. 2007. Tese (Doutorado) – Faculdade de Farmácia da Universidade Federal de Minas Gerais (UFMG), Belo Horizonte, 2007
- UNITED STATES ENVIRONMENTAL PROTECTION AGENCY (EPA). Method 1613. Tetra-through Octa-Chlorinated Dioxins and Furans by Isotope. Dilution HRGC/HRMS. U.S. rev. B, 1994.
- WENCLAWIAK, B.W.; KOCH, M.; HADJICOSTAS, E. Quality Assurance in Analytical Chemistry Training and Teaching 2nd Ed. New York: Springer, 2010, 331 p. ISBN 978-3-642-13608-5.
- WILLIAMS, A; MAGNUSSON, B. Eurachem/CITAC guide: Use of uncertainty information in compliance assessment (2nd ed. 2021). ISBN 978-0-948926-38-9. Available from www.eurachem.org.
- INTERNACIONAL UNION OF PURE AND APLLIED CHEMISTRY ORGANIZATION (IUPAC)/ISO/AOAC. Harmonised Guidelines for the Use of Recovery Information in Analytical Measurement. Pure and Applied Chemistry Vol. 71, pp. 337 – 348, 1999.
- EUROPEAN FEDERATION OF NATIONAL ASSOCIATIONS OF MEASUREMENT, TESTING AND ANALYTICAL LABORATORIES (EUROLAB). EUROLAB Technical Report 1/2006. Guide to the Evaluation of Measurement Uncertainty for Quantitative Test Results. 2006.
- EUROPEAN FEDERATION OF NATIONAL ASSOCIATIONS OF MEASUREMENT, TESTING AND ANALYTICAL LABORATORIES (EUROLAB). EUROLAB Technical Report 1/2007. Measurement Uncertainty revisited: Alternative approaches to uncertainty evaluation. 2007.
- INTERNACIONAL STANDARDS ORGANIZATION (ISO) ISSO IEC Guide 98-4 2012(E) - Uncertainty of Measurement -- Part 4, Role of measurement uncertainty in conformity assessment. 2012.
- LIRA, I.H. UND WÖGER, W. Evaluation of the uncertainty associated with a measurement result not corrected for systematic effects, Meas Sci Technol. 9, p. 1010-1011, 1998.
¶ 6. Disposições Gerais
As sugestões para aprimoramento ou possíveis correções deste documento devem ser direcionadas ao Departamento responsável, para alinhamento das melhores práticas de mercado, legislação vigente e/ou regulamentações, que não tenham sido contempladas na versão vigente.
¶ 7. Histórico de revisões
| Versão | Conteúdo alterado | Data | Motivo |
|---|---|---|---|
| 1 | - | 2011 | Elaboração do documento |
| 2 | 15.04.2025 | Revisão do documento |
¶ 8. Anexos
¶ ANEXO I - Definições
Ver item 1.1 deste manual.
¶ ANEXO II - Regressão Linear pelo Método dos Mínimos Quadrados Ordinários e Ponderados - Cálculos e Estudo de Caso
¶ A1 Cálculos dos parâmetros de regressão por MMQP
A equação regressão é dada pela equação a seguir,
Em que:
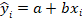
ŷi = variável dependente: é a resposta instrumental estimada (predita) pela equação de regressão no i-ésimo nível concentração.
a = estimativa da interseção, intercepto ou coeficiente angular.
b = estimativa da inclinação ou coeficiente angular.
xi = variável independente: são as concentrações conhecidas do analito nas soluções padrão de calibração em cada nível de concentração.
Procede-se à estimação de b, a e das somas quadráticas, considerando uma ponderação wi para o caso do MMQP:
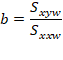
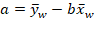
Em que as somas quadráticas Sxxw e Sxyw são calculadas pelas expressões:
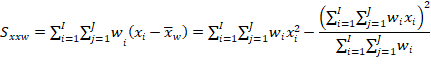
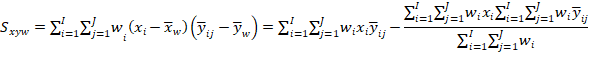
E as médias ponderadas de x e y são calculadas da seguinte forma:
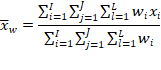
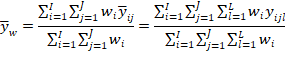
Em que yij médio é a média das L replicatas de injeção para um dado preparo de solução j e nível de concentração i e yijl é uma resposta instrumental para uma dada replica de leitura l, preparo de solução j e nível de concentração i.
Caso a aquisição das respostas instrumentais seja realizada sem replicata de leitura, desconsiderar o índice l e caso não seja relevante realizar diferentes preparos das soluções da curva, desconsiderar os índices j.
Todos os valores de yij médio têm o mesmo peso wi, que é usado para as J respostas instrumentais médias de cada nível.
Considerando os modelos mais utilizados, que dão maior ênfase nos níveis mais precisos e de menor concentração, podemos considerar as seguintes ponderações: Σsi2/Isi2, 1/x e 1/x2.
Caso o laboratório observe que os dados da resposta instrumental são homoscedásticos, ou seja interessante visualizar os parâmetros de regressão para o caso ordinário, para realizar os cálculos presentes neste estudo de caso basta alterar os valores de wi para 1 em todos os níveis, desta forma se obtém os parâmetros para o MMQO.
O coeficiente de determinação R2, calculado pela fórmula:
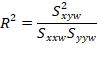
Em que Syyx, de modo análogo a Sxxw, pode ser calculado como:
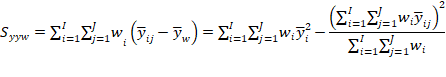
Como os parâmetros a e b são variáveis aleatórias, eles possuem desvios padrão associados, denominados aqui como s(a) e s(b). O cálculo desses desvios é realizado da seguinte forma:
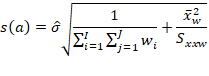
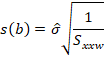
Em que σ^2 é definida como uma certa variância que multiplicada pelo fator de ponderação wi, estima a variância residual das médias de L replicatas de leitura em um dado nível i.
O valor de σ^2 pode ser obtido pela seguinte fórmula:
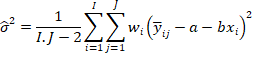
¶ A2 Cálculo das curvas de limite de previsão:
Com base no que foi descrito no tópico anterior sobre estimativa dos parâmetros de regressão, podemos concluir que o intercepto (a) e inclinação (b) são variáveis aleatórias. Isso deve-se ao fato de que a curva ajustada é proveniente de uma variável aleatória: a resposta instrumental Y.
Dito isso, é intuitivo dizer que os dados preditos pelo modelo ajustado terão componentes de erro associados tanto a resposta instrumental quanto aos parâmetros do modelo em si.
Portanto, as curvas de limite de previsão têm como objetivo estimar a região onde a maior parte dos resultados se encontra.
Define-se tal região/intervalo através de Limites de Previsão (LP) que podem ser calculados, em termos de da resposta instrumental (y), da seguinte forma:
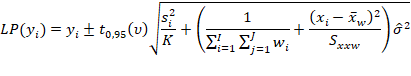
Em que t0,95(v) é o fator de Student para 95% de confiança e v graus de liberdade (IJ-2) para uma distribuição t bilateral e K é o número de replicações independentes no nível i (experimentos balanceados K = J)
Como o intuito é estimar o intervalo de predição para os resultados preditos x (concentrações), a equação acima pode ser reescrita da seguinte forma:
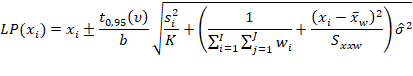
Vale ressaltar que o intervalo descrito acima abrange apenas as incertezas associadas aos parâmetros da curva (a e b) e resposta instrumental (y), e não abrange as demais incertezas decorrentes do processo de análise como um todo. Ou seja, este intervalo não compreende a incerteza da concentração do analito na amostra de ensaio.
Uma desvantagem do modelo descrito para estimar os LCs pelas equações acima é que os intervalos são calculados de modo discreto, ou seja, somente os I níveis que foram usados na elaboração da curva de calibração terão seus intervalos de predição estimados. Tal limitação está associada ao fata de que somente nos níveis i que foi possível estimar as variâncias si2. Qualquer outro valor de x diferente dos valores de xi não tem valores definidos para s2.
Tal característica é indesejada quando pretende-se predizer quaisquer valores de x em uma curva de calibração em uma dada faixa de trabalho.
Portanto, duas alternativas são propostas:
1ª: estimar através de uma outra regressão linear qual seria a relação funcional entre s2 e x, ou s e x. A partir daí, utilizar os parâmetros dessa regressão para estimar quaisquer valores de s2 dentro da faixa de trabalho da curva.
2ª: Escolher um modelo para os pesos wi tal que wi tenha uma relação funcional bem descrita com a concentração x, dentro da faixa de trabalho. Os modelos de wi mais comuns, até mesmo em softwares das principais marcas de espectrômetros, são: wi = 1/x ou 1/x2 ou 1/y ou 1/y2.
Em termos de cálculos, a equação anterior pode ser reescrita como definido por Massart [51]:
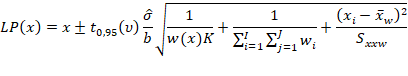
Em que w(x) é uma função conhecida dependente de x.
¶ A3 Análise da qualidade do ajuste:
Agora, faz-se necessária a avaliação se os ajustes propostos considerando os pesos escolhidos, de fato são realmente confiáveis para realizar as predições necessárias.
Existem diversas ferramentas para avaliação da qualidade de um ajuste disponíveis na literatura e em outros guias de validação ao redor do mundo. Porém, neste manual, recomenda-se o uso da Recuperação da concentração retrocalculada (Rcr (%)) conforme discutido no item II.2.2 e o teste de falta de ajuste através de análise de variância (ANOVA) para o caso de resultados heteroscedásticos.
A seguir, a Tabela 1 traz os cálculos para realizar o teste de falta de ajuste da ANOVA [51]:
Tabela 1: Análise de variância (ANOVA) para avaliação da falta de ajuste em curvas de calibração com respostas instrumentais heteroscedásticas.

p-valores acima de 0,05 indicam a aceitação da hipótese nula de que não há evidência estatística para falta de ajuste, ou seja, o ajuste foi adequado aos dados.
Exemplo: Dados reais de uma curva de calibração de aflatoxina M1 em leite cru bovino adquirida em sistema LC-MS/MS com ionização ESI e analisador de massas do tipo triplo quadrupolo, sem a utilização de padrão interno. A Tabela 2 a seguir mostra os dados de concentração e resposta instrumental (área cromatográfica), para os I níveis, J réplicas independentes e L réplicas de injeção.
Tabela 2: Resultados obtidos para uma curva de calibração de aflatoxina M1 em leite obtida em sistema LC-MS/MS (QQQ).
|
Nível |
Preparo |
Conc. xi (mg/L) |
Área cromatográfica |
|||
|
Inj. l = 1 |
Inj. l = 2 |
Inj. l = 3 |
|
|||
|
i = 1 |
j = 1 |
0,0266 |
1112 |
915 |
1031 |
1019 |
|
j = 2 |
0,0266 |
830 |
1036 |
694 |
853 |
|
|
j = 3 |
0,0266 |
1284 |
1029 |
690 |
1001 |
|
|
i = 2 |
j = 1 |
0,0531 |
2273 |
1702 |
2230 |
2068 |
|
j = 2 |
0,0531 |
1515 |
1698 |
1829 |
1681 |
|
|
j = 3 |
0,0531 |
1896 |
1704 |
2066 |
1889 |
|
|
i = 3 |
j = 1 |
0,1054 |
3270 |
3154 |
3189 |
3204 |
|
j = 2 |
0,1054 |
3166 |
2354 |
3157 |
2892 |
|
|
j = 3 |
0,1054 |
4041 |
4112 |
3365 |
3839 |
|
|
i = 4 |
j = 1 |
0,2138 |
6408 |
7069 |
6677 |
6718 |
|
j = 2 |
0,2138 |
4963 |
5228 |
4861 |
5017 |
|
|
j = 3 |
0,2138 |
7997 |
7975 |
7761 |
7911 |
|
|
i = 5 |
j = 1 |
0,4250 |
14260 |
14210 |
13140 |
13870 |
|
j = 2 |
0,4250 |
11450 |
12560 |
11290 |
11767 |
|
|
j = 3 |
0,4250 |
13080 |
16050 |
16790 |
15307 |
|
|
i = 6 |
j = 1 |
0,8500 |
24140 |
28230 |
31140 |
27837 |
|
j = 2 |
0,8500 |
25350 |
21870 |
23340 |
23520 |
|
|
j = 3 |
0,8500 |
28270 |
28890 |
30580 |
29247 |
|
|
i = 7 |
j = 1 |
1,7000 |
57240 |
56730 |
51640 |
55203 |
|
j = 2 |
1,7000 |
41530 |
43790 |
43980 |
43100 |
|
|
j = 3 |
1,7000 |
65120 |
61600 |
68380 |
65033 |
|
Tabela 3: Parâmetros de regressão estimados com diferentes pesos para a curva de calibração de aflatoxina M1 em leite.
|
Parâmetro |
Pesos wi |
|||
|
Σsi2/Isi2 |
1/x |
1/x2 |
1 (MMQO) |
|
|
Sxxw |
11,5355 |
8,1541 |
12,1271 |
6,6819 |
|
Syyw |
1,1740E+10 |
8,4053E+09 |
1,2230E+10 |
7,0953E+09 |
|
Sxyw |
3,6346E+05 |
2,5855E+05 |
3,7962E+05 |
2,1354E+05 |
|
xw |
0,03554 |
0,09370 |
0,03959 |
0,48199 |
|
yw |
1250,3 |
3067,9 |
1374,2 |
15379,9 |
|
a |
130,5 |
97,0 |
134,9 |
-23,4 |
|
b |
31508 |
31708 |
31303 |
31958 |
|
σ^2 |
1,5164E+07 |
1,0903E+07 |
1,8241E+07 |
1,4264E+07 |
|
s(a) |
58,1 |
245,7 |
74,7 |
1084,1 |
|
s(b) |
1147 |
1156 |
1226 |
1461 |
|
R2 |
0,9755 |
0,9754 |
0,9717 |
0,9618 |
Observando a Tabela 3 com os resultados obtidos para os parâmetros com diferentes ponderações na construção da curva de calibração, os desvios padrão do intercepto a e inclinação b, s(a) e s(b), respectivamente, não foram muito distintos, exceto para o caso da regressão ordinária (MMQO), que apresentou os maiores desvios padrão.
A seguir, continuando os exemplos, as curvas de limite de previsão serão calculadas com base no modelo de ponderação 1/x, pois este modelo, além de ser contínuo, ajusta bem aos resultados e é de fácil uso nos laboratórios devido a disponibilidade dessa ponderação na maioria dos softwares, conforme é apresentado na tabela 4 e na figura 1.
Tabela 4: Limites de previsão para o modelo de ponderação 1/x utilizado na curva de calibração de aflatoxina M1 em leite
|
Concentração xi (mg/L) |
ŷi |
Limites de previsão (95%) |
|
|
Inferior |
Superior |
||
|
0,0266 |
940 |
126 |
1755 |
|
0,0531 |
1781 |
747 |
2814 |
|
0,1054 |
3439 |
2064 |
4815 |
|
0,2138 |
6876 |
4952 |
8800 |
|
0,4250 |
13573 |
10812 |
16334 |
|
0,8500 |
27049 |
22914 |
31184 |
|
1,7000 |
54001 |
47490 |
60512 |
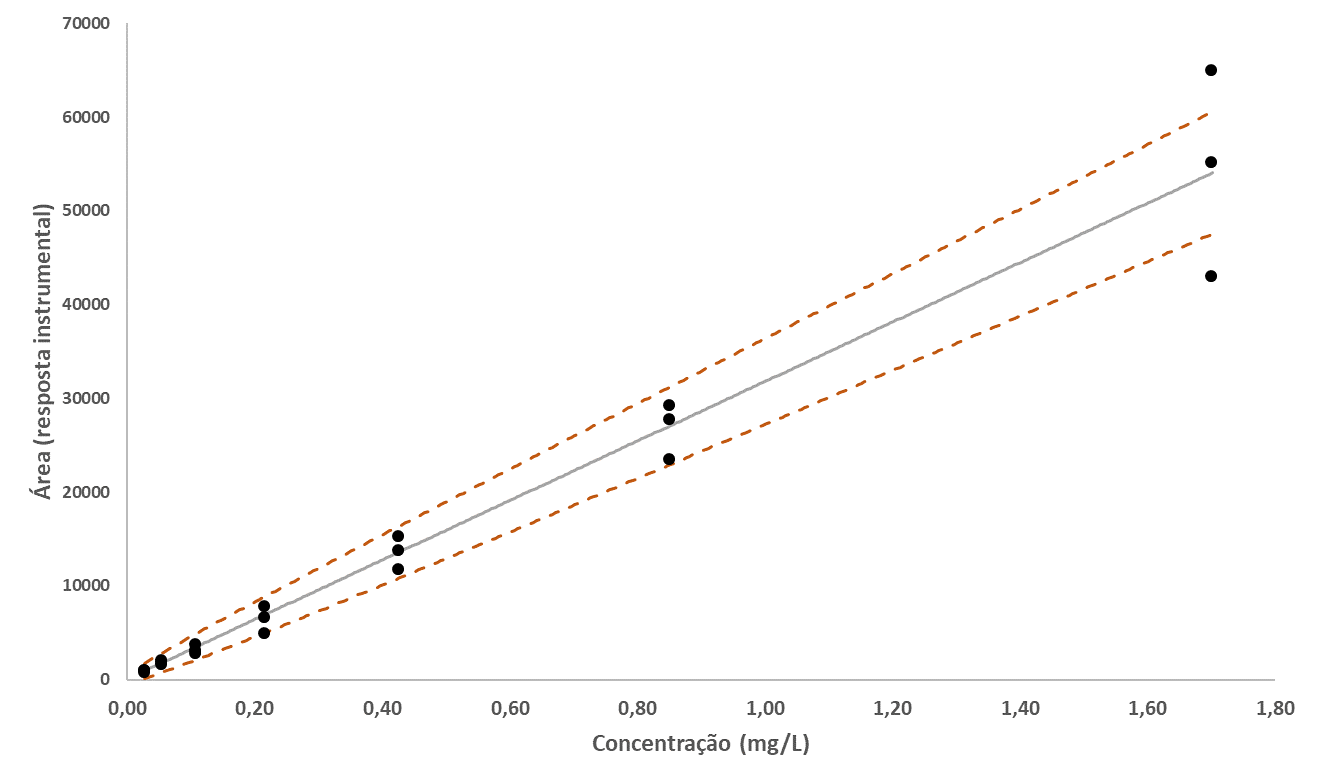
Por fim, necessita-se avaliar os modelos propostos se atendem ao propósito de predição de resultados, ou seja, necessita-se de avaliar a qualidade do(s) ajuste(s)
A Tabela 5 a seguir traz os resultados das concentrações retrocalculadas expressas como Recuperação (% Rcr), que é a razão entre a concentração obtida pela predição e a concentração nominal xi. Os resultados foram trazidos para cada replicata independente j e para 4 diferentes pesos wi, incluindo o caso ordinário (wi = 1).
Tabela 5: Resultados da avaliação residual dos ajustes, através dos valores de Recuperação das concentrações retrocalculadas, considerando os 4 diferentes pesos wi obtidos para a curva de calibração de aflatoxina M1 em leite.
|
Conc. (mg/L) |
|
peso wi = Σsi2/Isi2 |
peso wi = 1/x |
peso wi = 1/x2 |
peso wi = 1 |
||||
|
wi |
%Rcr |
wi |
%Rcr |
wi |
%Rcr |
wi |
%Rcr |
||
|
0,0266 |
1019 |
2331 |
106 |
37,6 |
109 |
1413 |
106 |
1 |
123 |
|
0,0266 |
853 |
2331 |
86 |
37,6 |
90 |
1413 |
86 |
1 |
103 |
|
0,0266 |
1001 |
2331 |
104 |
37,6 |
107 |
1413 |
104 |
1 |
121 |
|
0,0531 |
2068 |
513 |
116 |
18,8 |
117 |
355 |
116 |
1 |
123 |
|
0,0531 |
1681 |
513 |
93 |
18,8 |
94 |
355 |
93 |
1 |
100 |
|
0,0531 |
1889 |
513 |
105 |
18,8 |
106 |
355 |
106 |
1 |
113 |
|
0,1054 |
3204 |
82,9 |
93 |
9,5 |
93 |
90,0 |
93 |
1 |
96 |
|
0,1054 |
2892 |
82,9 |
83 |
9,5 |
84 |
90,0 |
84 |
1 |
87 |
|
0,1054 |
3839 |
82,9 |
112 |
9,5 |
112 |
90,0 |
112 |
1 |
115 |
|
0,2138 |
6718 |
9,13 |
98 |
4,7 |
98 |
21,9 |
98 |
1 |
99 |
|
0,2138 |
5017 |
9,13 |
73 |
4,7 |
73 |
21,9 |
73 |
1 |
74 |
|
0,2138 |
7911 |
9,13 |
115 |
4,7 |
115 |
21,9 |
116 |
1 |
116 |
|
0,4250 |
13870 |
6,09 |
103 |
2,4 |
102 |
5,5 |
103 |
1 |
102 |
|
0,4250 |
11767 |
6,09 |
87 |
2,4 |
87 |
5,5 |
87 |
1 |
87 |
|
0,4250 |
15307 |
6,09 |
113 |
2,4 |
113 |
5,5 |
114 |
1 |
113 |
|
0,8500 |
27837 |
2,17 |
103 |
1,2 |
103 |
1,4 |
104 |
1 |
103 |
|
0,8500 |
23520 |
2,17 |
87 |
1,2 |
87 |
1,4 |
88 |
1 |
87 |
|
0,8500 |
29247 |
2,17 |
109 |
1,2 |
108 |
1,4 |
109 |
1 |
108 |
|
1,7000 |
55203 |
0,16 |
103 |
0,6 |
102 |
0,3 |
103 |
1 |
102 |
|
1,7000 |
43100 |
0,16 |
80 |
0,6 |
80 |
0,3 |
81 |
1 |
79 |
|
1,7000 |
65033 |
0,16 |
121 |
0,6 |
120 |
0,3 |
122 |
1 |
120 |
Observando a Tabela 5 acima, nota-se que o modelo dos mínimos quadrados ordinários (MMQO) apresentou maiores valores de resíduos relativos no nível mais baixo da curva de calibração. Tal fato comumente ocorre quando resultados heteroscedásticos de variância crescente com a concentração são ajustados por MMQO, pois a contribuição alta dos níveis superiores acaba deslocando a curva nos níveis mais baixos, algo que é minimizado quando se usa uma ponderação mais adequada no modelo.
A seguir, a Figura 2 abaixo traz um gráfico de dispersão mostrando a distribuição dos resíduos da regressão obtida com ponderação 1/x. As demais ponderações apresentam perfis muito similares. O relevante neste diagrama é mostrar o comportamento heterocedástico dos resíduos. Tal comportamento é bastante comum em técnicas espectrométricas para análises de resíduos e contaminantes.
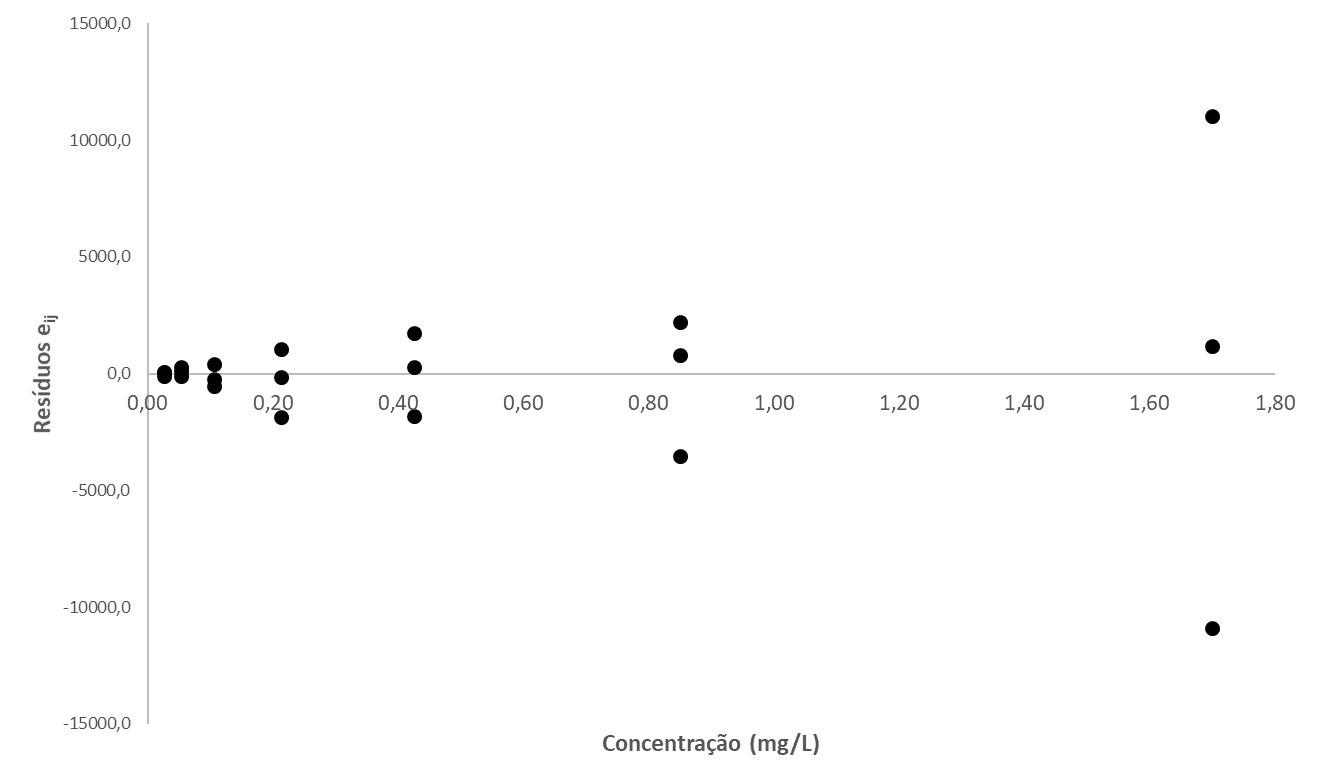
Além da análise de resíduos, o teste de falta de ajuste por ANOVA é uma boa ferramenta para avaliação do ajuste do modelo proposto.
A Tabela 6 a seguir traz os resultados obtidos do teste de falta de ajuste por ANOVA ponderada para a curva de aflatoxina M1 em leite, utilizando 4 tipos diferentes de ponderação, incluindo o MMQO.
Tabela 6: Resultados da análise de falta de ajusta por ANOVA para 4 tipos de ponderação para a curva de calibração de aflatoxina M1 em leite.
|
Fator |
GL |
Peso wi |
SQ |
MQ |
Fcalc |
p-valor |
|
Falta de ajuste |
5 |
Σsi2/Isi2 |
1,780E+07 |
3,559E+06 |
0,18433 |
0,96384 |
|
1/x |
3,051E+06 |
6,101E+05 |
0,04185 |
0,99877 |
||
|
1/x2 |
1,874E+07 |
3,749E+06 |
0,16009 |
0,97317 |
||
|
1 |
6,919E+05 |
1,384E+05 |
0,00717 |
0,99998 |
||
|
Erro puro |
14 |
Σsi2/Isi2 |
2,703E+08 |
1,931E+07 |
|
|
|
1/x |
2,041E+08 |
1,458E+07 |
|
|
||
|
1/x2 |
3,278E+08 |
2,342E+07 |
|
|
||
|
1 |
2,703E+08 |
1,931E+07 |
|
|
Neste exemplo, pode ser notado na tabela acima que todos os p-valores foram muito superiores a 0,05, indicando que todos os modelos apresentaram um bom ajuste, com base no teste de falta de ajuste da ANOVA. Entretanto, mesmo que o caso de MMQO tenha apresentado o p-valor mais alto, indicando assim um melhor ajuste, este modelo não deve ser usado para os casos em que os resíduos de regressão apresentam comportamento heterocedástico. Aplicando um modelo equivocado, os testes de hipótese utilizados não terão significância/confiança compatível com o declarado no início do teste, prejudicando drasticamente o poder do teste e consequente tomada de decisão.
¶ ANEXO III - Impacto do uso do MMQO e do MMQP para um mesmo conjunto de dados: Estudo de caso
Exemplo: Estimar regressão linear por MMQO e MMQP (Σsi2/Isi2) para os dados da tabela abaixo referente ao estudo de linearidade do método de determinação de resíduos de agrotóxicos em frutas por HPLC-MS/MS.
Tabela 1: Valores obtidos para a avaliação da linearidade.
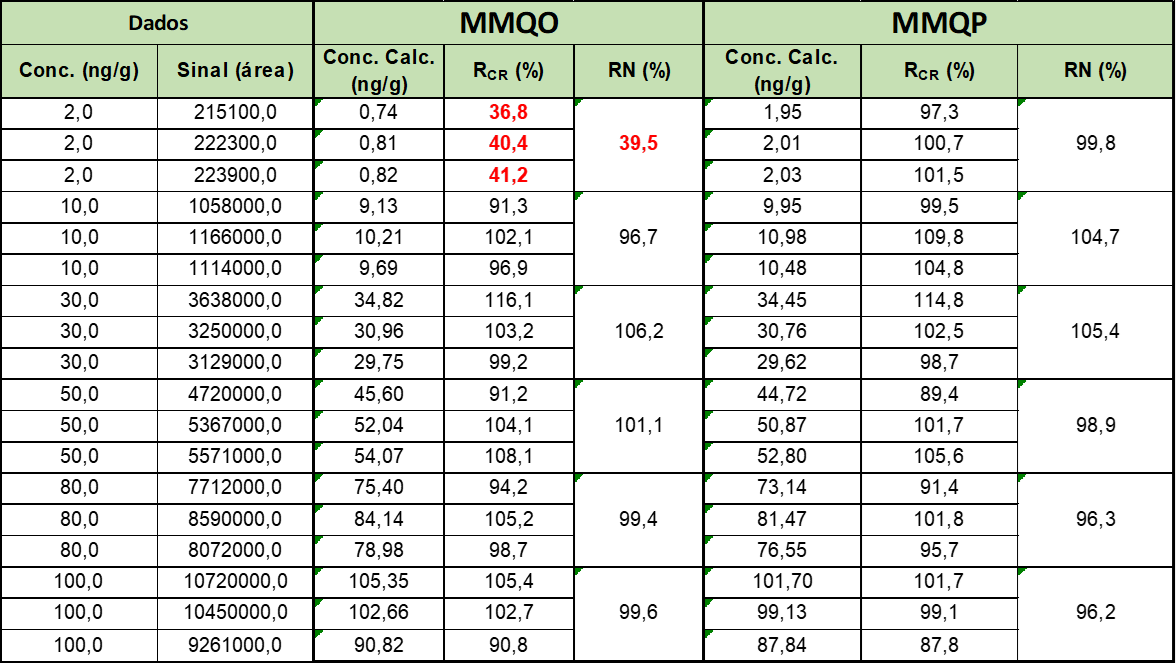
A partir dos dados acima foram plotadas as duas curvas de calibração conforme a figura 1 abaixo:
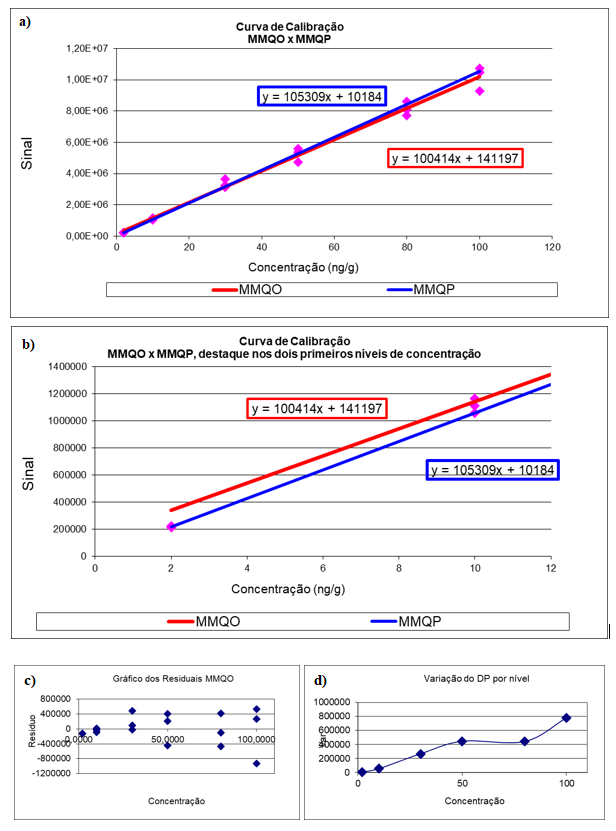
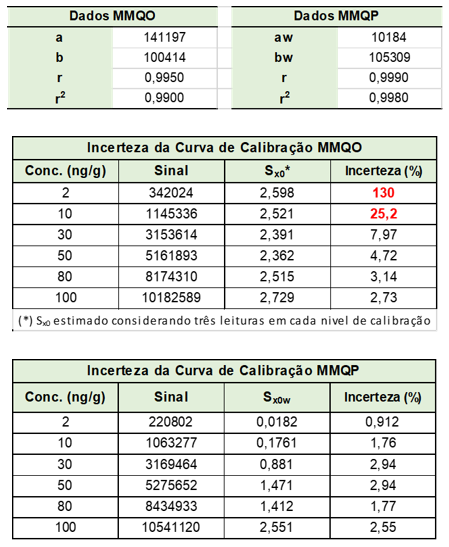
Observe as evidências da falta de ajuste por MMQO, bem como a melhor adequação ao modelo MMQP ao avaliar:
- Gráfico dos resíduos, visualmente demonstrando a heteroscedasticidade, já que o desvio padrão aumenta na medida que o valor de x se eleva, fato reforçado pelo gráfico do desvio padrão do sinal vs nível de calibração;
- Ajuste linear do gráfico, em especial nos dois primeiros níveis de calibração, demonstrando como regressão MMQP está mais bem ajustada aos pontos de menor concentração;
- Os valores de Recuperação da concentração retrocalculada (Rcr (%)) por ponto e por Nível (RN (%)), os quais estão bem mais adequados para o ajuste por MMQP;
- Valores de Incerteza obtidos para cada modelo, por nível de concentração, destacando-se os valores inadequados (em vermelho) obtidos pelo modelo MMQO;
- Observar os valores de coeficientes de correlação (r e r2) acima e 0,99 para ambos os métodos, demonstrando que os coeficientes de correlação sozinhos não são bons indicativos do da adequação do ajuste linear.
¶ ANEXO IV - Teste de Youden
Uso do planejamento fatorial fracionário conhecido como Teste de Youden é realizado por meio de um delineamento que envolve sete parâmetros analíticos combinados em oito experimentos.
Selecionar os fatores a serem avaliados, seus valores nominais (descritos no método) e os respectivos valores de variação, preenchendo uma tabela de acordo com o exemplo:
Tabela 1: Fatores a serem avaliados e os valores de variação
|
Fator |
Nominal |
Variação |
|
Massa de amostra (g) |
5,00 |
7,50 |
|
Volume de HNO3(concentrado) (mL) |
15,0 |
12,0 |
|
Volume de HCl(concentrado) (mL) |
5,0 |
4,0 |
|
Temperatura de aquecimento (°C) |
100 |
120 |
|
Tempo de aquecimento (h) |
1,0 |
1,5 |
|
Contêiner |
Cadinho |
Béquer |
|
Agitação |
Sim |
Não |
Executar o ensaio com as combinações da tabela abaixo em duplicata e aleatoriamente, inclusive entre as replicatas de cada combinação.
Tabela 2: Combinação de ensaios.
|
Valor do fator |
Combinação para os ensaios |
|||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
|
A/a |
A |
A |
A |
A |
a |
a |
a |
a |
|
B/b |
B |
B |
b |
b |
B |
B |
b |
b |
|
C/c |
C |
c |
C |
c |
C |
c |
C |
c |
|
D/d |
D |
D |
d |
d |
d |
d |
D |
D |
|
E/e |
E |
e |
E |
e |
e |
E |
e |
E |
|
F/f |
F |
f |
f |
F |
F |
f |
f |
F |
|
G/g |
G |
g |
g |
G |
g |
G |
G |
g |
|
Resultado Observado |
S |
T |
U |
V |
W |
X |
Y |
Z |
Calcular a média e a variância (s2) para cada combinação de ensaio.
Para determinar o efeito da variação de um fator, encontrar os 4 valores correspondentes às letras maiúsculas e subtrair dos 4 valores correspondentes às letras minúsculas, segundo a equação:
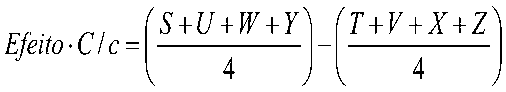
Calcular o efeito para todos os fatores e construir um gráfico de barras horizontais com os resultados, usando o módulo dos valores.
Estimar a variância de todos os efeitos empregando a equação:
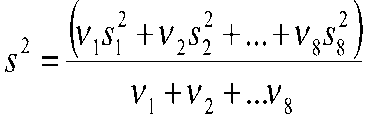
Onde:
i = ni – 1 é o número de graus de liberdade de si2, sendo si2 a estimativa da variância do i-ésimo ensaio.
Calcular o erro padrão do efeito aplicando a equação:
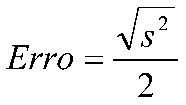
Multiplicar o valor do erro puro por 2,306 (valor do t de Student para 8 graus de liberdade com 95% de confiança, realizados em duplicata) para traçar a linha de significância dos efeitos no gráfico de barras.
Calcular se há diferença estatística significativa (95%) entre os valores dos resultados obtidos para maiúsculas e minúsculas, empregando o teste F.
Calcular o desvio-padrão dos efeitos (sDi) pela fórmula:
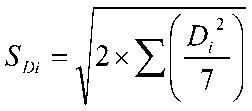
Onde:
D1 é valor do efeito (ou diferença).
¶ ANEXO V - Modelo de Relatório de Validação de Método
Título do Método
DECLARAÇÃO DE CONFORMIDADE ANALÍTICA
Declaramos que o estudo aqui apresentado foi executado da exata maneira descrita neste relatório, sendo os resultados obtidos reprodutíveis. Estes resultados refletem fielmente as conclusões aqui apresentadas.
Todos os rascunhos e dados brutos obtidos encontram-se disponíveis e arquivados, sob as codificações internas de nossos registros, com rastreabilidade garantida pelo sistema de gestão da qualidade.
Este relatório foi elaborado e redigido pelos responsáveis abaixo descritos que se responsabilizam pelos dados aqui apresentados.
| Aprovação do método: | Sim | Não | ||||||
| Responsável(eis) pelo estudo: | ||||||||
| Assinatura(s): | Data: | |||||||
| Responsável(eis) pela elaboração do relatório: | ||||||||
| Assinatura (s): | Data: | |||||||
|
|
||||||||
| Responsável Técnico pela Unidade Laboratorial: | ||||||||
| Assinatura: | Data: | |||||||
1. INTRODUÇÃO
2. OBJETIVO
3. MATERIAL E MÉTODO
Descrição detalhada do método, caso seja um procedimento já em uso, citar o procedimento.
Todas as informações referentes aos materiais e métodos para o preparo de soluções, execução do método de extração e dimensionamento dos parâmetros cromatográficos e de detecção.
Rastreabilidade dos reagentes, soluções e padrões
|
Nome |
Lote |
Fabricante |
Grau de pureza (%) |
Validade |
Rastreabilidade dos instrumentos e vidrarias utilizados
|
Nome |
Identificação do Instrumento |
Rastreabilidade dos demais materiais utilizados
As soluções padrão, reagentes, solventes e equipamentos utilizados durante a validação do método
Os analitos propostos à validação.
4. VALIDAÇÃO INTRALABORATORIAL
Descrever os parâmetros avaliados, de que forma foi feita a avaliação, número de amostras analisadas em cada etapa, os cálculos envolvidos e os critérios de aceitabilidade utilizados, abaixo uma descrição dos tópicos que normalmente são avaliados, podendo estar presente outros assuntos conforme for a necessidade
4.1 LINEARIDADE
Como foi construída a curva de calibração, qual tipo de curva (CCS, CCM, CCF), quantidade de níveis e réplicas por nível, sequência de injeção. Como é avaliada a linearidade, quais testes são realizados, quais critérios de aceitabilidade.
4.2 EFEITO MATRIZ
Como foi realizado o teste de efeito matriz, como foi feita a comparação, qual critério de aceitabilidade.
4.3 REPETIBILIDADE E REPRODUTIBILIDADE (VERACIDADE E PRECISÃO)
Quais níveis foram avaliados, quantas repetições por nível. Para a reprodutibilidade quantos lotes foram utilizados, quais os fatores variaram (dia, analista, equipamento...). Quais critérios para aceitabilidade.
4.4 LIMITE DE DETECÇÃO (LD), LIMITE DE QUANTIFICAÇÃO (LQ), LIMITE DE DECISÃO (CCα) CAPACIDADE DE DETECÇÃO (CCβ)
Como foram estabelecidos os limites, quando aplicáveis.
4.5 SELETIVIDADE
Como foi estabelecido e quais critérios para aceitabilidade.
4.6 RAZÃO DE ÍONS
Como foi estabelecido e quais critérios para aceitabilidade.
4.7 - CÁLCULO DA INCERTEZA
Como foi estimada a incerteza inicial do método, por qual procedimento matemático.
5. RESULTADOS E DISCUSSÕES
Apresentar os resultados aplicáveis, preferencialmente em forma de tabela, conforme os exemplos abaixo.
Exemplo: Tabela com os resultados da avaliação da linearidade:

Exemplo: Tabela Resultados de Precisão (repetibilidade e reprodutibilidade), Veracidade / Recuperação, LD, LQ, CCα, CCβ, Incerteza, Razão de Íons.

Resultados com valores fora dos critérios de aceitabilidade devem ser destacados e discutidos.
5.1 LINEARIDADE
5.2 EFEITO MATRIZ
5.3 REPETIBILIDADE E REPRODUTIBILIDADE (EXATIDÃO E PRECISÃO)
5.3.1 VERACIDADE (RECUPERAÇÃO)
5.3.2 REPETIBILIDADE E REPRODUTIBILIDADE (PRECISÃO)
5.4 LIMITE DE DETECÇÃO (LD), LIMITE DE QUANTIFICAÇÃO (LQ), LIMITE DE DECISÃO (CCα) CAPACIDADE DE DETECÇÃO (CCβ)
5.5 SELETIVIDADE
5.6 RAZÃO DE ÍONS
5.7 CÁLCULO DA INCERTEZA
6. CONCLUSÃO
Conclusão quanto à adequação do método ao proposito para o escopo final.
Exemplo: Tabela com os analitos validados (escopo) e os limites necessários, e.g.: LQ, e Incerteza, quando aplicável.

7. REFERÊNCIAS
Documentos utilizados na validação, tais como: artigos científicos, guias de validação, métodos oficiais, referências normativas...
8. ANEXOS